
Để sử dụng toàn bộ tiện ích nâng cao của Hệ Thống Pháp Luật vui lòng lựa chọn và đăng ký gói cước.
| BỘ Y TẾ | CỘNG HÒA XÃ HỘI CHỦ NGHĨA VIỆT NAM |
| Số: 658/QĐ-QLD | Hà Nội, ngày 19 tháng 9 năm 2023 |
VỀ VIỆC BAN HÀNH QUY TRÌNH THAO TÁC CHUẨN TRONG HỆ THỐNG QUẢN LÝ CHẤT LƯỢNG THEO TIÊU CHUẨN ISO 9001:2015 ÁP DỤNG VÀO HOẠT ĐỘNG QUẢN LÝ NHÀ NƯỚC TẠI CỤC QUẢN LÝ DƯỢC
CỤC TRƯỞNG CỤC QUẢN LÝ DƯỢC
Căn cứ Nghị định số 95/2022/NĐ-CP ngày 15/11/2022 của Chính phủ quy định chức năng, nhiệm vụ, quyền hạn và cơ cấu tổ chức của Bộ Y tế;
Căn cứ Quyết định số 1969/QĐ-BYT ngày 26/4/2023 của Bộ trưởng Bộ Y tế quy định chức năng, nhiệm vụ, quyền hạn và cơ cấu tổ chức của Cục Quản lý Dược thuộc Bộ Y tế;
Căn cứ Quyết định số 388/QĐ-QLD ngày 29/5/2023 của Cục trưởng Cục Quản lý Dược về việc ban hành Quy định chức năng, nhiệm vụ, quyền hạn của Văn phòng và các phòng thuộc Cục Quản lý Dược;
Căn cứ yêu cầu thực tế công tác quản lý của Cục Quản lý Dược;
Theo đề nghị của Chánh Văn phòng Cục.
QUYẾT ĐỊNH:
Điều 1. Ban hành kèm theo Quyết định này 01 Quy trình thao tác chuẩn trong Hệ thống quản lý chất lượng theo tiêu chuẩn ISO 9001:2015 áp dụng vào hoạt động quản lý nhà nước tại Cục Quản lý Dược, cụ thể:
Quy trình giải quyết hồ sơ đăng ký thay đổi, bổ sung thuốc, nguyên liệu làm thuốc trong quá trình lưu hành (mã số QT.ĐK.08.08 thay thế quy trình mã số QT.ĐK.08.07).
Điều 2. Quyết định này có hiệu lực kể từ ngày ký.
Điều 3. Các Ông/bà: Lãnh đạo Cục, Lãnh đạo Ban QMS, Chánh Văn phòng, Trưởng các phòng thuộc Cục Quản lý Dược chịu trách nhiệm thi hành Quyết định này./.
|
| CỤC TRƯỞNG |
MÃ SỐ: QT.ĐK.08.08
QUY TRÌNH
GIẢI QUYẾT HỒ SƠ ĐĂNG KÝ THAY ĐỔI/BỔ SUNG THUỐC, NGUYÊN LIỆU LÀM THUỐC TRONG QUÁ TRÌNH LƯU HÀNH
1. MỤC ĐÍCH
Quy trình này nhằm quy định thống nhất việc tiếp nhận, thẩm định và giải quyết hồ sơ thay đổi, bổ sung giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc (bao gồm thuốc hóa dược, thuốc dược liệu, vắc xin, sinh phẩm, thuốc phóng xạ, nguyên liệu làm thuốc) sản xuất trong nước, sản xuất tại nước ngoài trong thời hạn giấy đăng ký lưu hành còn hiệu lực.
2. PHẠM VI ÁP DỤNG
- Quy trình này áp dụng đối với hồ sơ đăng ký thay đổi, bổ sung thuốc, nguyên liệu làm thuốc sản xuất trong nước, sản xuất tại nước ngoài đã có giấy đăng ký lưu hành trong thời hạn giấy đăng ký lưu hành còn hiệu lực (trừ hoạt động đăng ký thay đổi, bổ sung giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc đối với nội dung thay đổi nhỏ chỉ yêu cầu thông báo).
- Các cán bộ, chuyên viên và người được phân công tham gia quá trình tiếp nhận, xử lý và giải quyết hồ sơ thay đổi, bổ sung giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc.
3. TÀI LIỆU THAM CHIẾU
- Luật Dược số 105/2016/QH13 ngày 06/4/2016.
- Thông tư số 08/2022/TT-BYT ngày 05/9/2022 của Bộ Y tế quy định việc đăng ký lưu hành thuốc, nguyên liệu làm thuốc;
- Thông tư số 05/2010/TT-BYT ngày 01/3/2010 của Bộ Y tế hướng dẫn bảo mật dữ liệu trong ĐKT.
- Thông tư số 16/2023/TT-BYT ngày 15/8/2023 của Bộ Y tế quy định việc đăng ký lưu hành đối với thuốc gia công, thuốc chuyển giao công nghệ tại Việt Nam.
- Chủ trương Hội đồng tư vấn cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc đợt 110.3 thuốc nước ngoài ngày 28/10/2022.
4. TRÁCH NHIỆM THỰC HIỆN
- Lãnh đạo phòng ĐKT có trách nhiệm thường xuyên kiểm tra, đôn đốc, tổ chức thực hiện theo đúng quy định tại quy trình này.
- Lãnh đạo Cục, Lãnh đạo phòng ĐKT và chuyên viên có liên quan của phòng ĐKT, VPC có trách nhiệm thực hiện và tuân thủ những quy định trong quy trình này.
- Lãnh đạo các phòng có liên quan trong Cục QLD có trách nhiệm phối hợp, kiểm tra và bảo đảm những quy định trong quy trình này được thực hiện và tuân thủ.
- Phòng Đăng ký thuốc chịu trách nhiệm phối hợp với Văn phòng Hội đồng tổ chức họp HĐ và xử lý biên bản họp Hội đồng.
5. ĐỊNH NGHĨA VÀ CHỮ VIẾT TẮT
5.1. Thuật ngữ:
- Chuyên viên phụ trách: là chuyên viên được giao nhiệm vụ thụ lý hồ sơ đăng ký thuốc trong nước, thuốc nhập khẩu, vắc xin, sinh phẩm theo danh sách phân công của Lãnh đạo Phòng.
- Chuyên viên đầu mối: là chuyên viên được giao thực hiện chức năng, nhiệm vụ cụ thể trong bảng phân công công việc của Lãnh đạo phòng liên quan đến các bước trong quy trình.
- Hội đồng: là Hội đồng tư vấn cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc được thành lập theo Quyết định của Bộ trưởng Bộ Y tế.
- Đơn vị thẩm định: là đơn vị nhận hồ sơ thay đổi, bổ sung vắc xin, sinh phẩm từ Cục QLD để tiến thành thẩm định tại đơn vị, bao gồm: Trung tâm dược lý lâm sàng - Trường đại học Y Hà Nội, Viện kiểm định vắc xin và sinh phẩm y tế.
- Danh mục hồ sơ trình Hội đồng: bao gồm 03 danh mục hồ sơ đã hoàn thành thẩm định của các doanh nghiệp để trình hội đồng, bao gồm: (1) hồ sơ được thẩm định đạt yêu cầu, (2) hồ sơ được thẩm định không đạt yêu cầu, (3) hồ sơ đã thẩm định cần xin ý kiến Hội đồng.
- Hồ sơ thay đổi, bổ sung phải trình Hội đồng: bao gồm các hồ sơ thay đổi lớn về chỉ định, liều dùng, đối tượng dùng thuốc, phân loại biệt dược gốc, sinh phẩm tham chiếu (theo MiV-PA38), thuốc có báo cáo nghiên cứu tương đương sinh học (theo MiV-PA37); các trường hợp thay đổi, bổ sung khác cần xin ý kiến Hội đồng.
- Lãnh đạo Phòng Đăng ký thuốc: là Trưởng phòng hoặc Phó trưởng phòng được Cục trưởng phân công phụ trách nhóm một trong ba nhóm: nhóm thuốc sản xuất trong nước; nhóm thuốc nhập khẩu; nhóm vắc xin, sinh phẩm.
5.2. Chữ viết tắt:
- BBTĐ: Biên bản thẩm định;
- BB: Biên bản
- BM: Biểu mẫu.
- BE hoặc BA/BE: Tương đương sinh học
- BS: Bổ sung
- CV: Công văn
- CGTĐ: Chuyên gia thẩm định hồ sơ đăng ký thay đổi, bổ sung;
- CVPT: Chuyên viên phụ trách doanh nghiệp.
- DN: Doanh nghiệp
- ĐKT: Đăng ký thuốc
- GĐKLH: Giấy đăng ký lưu hành
- HDSD: Hướng dẫn sử dụng
- HĐ: Hội đồng tư vấn cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc/VX, sinh phẩm.
- HSTĐ/BS: Hồ sơ đăng ký thay đổi, bổ sung (trừ hồ sơ đăng ký thay đổi nhỏ chỉ yêu cầu thông báo).
- HSBS: Hồ sơ bổ sung của hồ sơ đăng ký thay đổi, bổ sung.
- HS: Hồ sơ
- HSLS: Hồ sơ lâm sang
- LĐ: Lãnh đạo
- LĐP.ĐKT: Lãnh đạo phòng Đăng ký thuốc
- LV: làm việc
- NICVB: Viện Kiểm định Quốc gia Vắc xin - Sinh phẩm y tế
- P. ĐKT: Phòng Đăng ký thuốc.
- QCĐG: Quy cách đóng gói
- SĐK: Số đăng ký
- SX: Sản xuất
- TĐHS: thẩm định hồ sơ
- VPC: Văn phòng Cục
- VPHĐ: Văn phòng Hội đồng tư vấn cấp Giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc
- YC: Yêu cầu;
6. QUY ĐỊNH CHUNG
6.1. Một số quy định chung:
6.1.1. Quy trình tiếp nhận, thẩm định hồ sơ thay đổi/bổ sung và trình Hội đồng đối với hồ sơ thay đổi, bổ sung giấy đăng ký lưu hành được thống nhất áp dụng và thực hiện chung cho tất cả các bộ phận có liên quan của Cục QLD. Trường hợp nội dung tương ứng quy định trong các quy trình tham chiếu khác với nội dung quy định trong quy trình này thì thống nhất áp dụng theo quy định của quy trình này.
6.1.2. Quy trình này áp dụng để xử lý cho các trường hợp nộp hồ sơ trực tuyến theo quy định của Thông tư số 08/2022/TT-BYT ngày 05/9/2022 của Bộ Y tế quy định việc đăng ký lưu hành thuốc, nguyên liệu làm thuốc.
6.1.3. Đối với hồ sơ đã nộp bản giấy mà chưa được phê duyệt nội dung đề nghị thay đổi/ bổ sung: tiếp tục thực hiện theo quy trình số QT.ĐK.08.07. Trường hợp có Quyết định công bố triển khai nộp hồ sơ trực tuyến, các hồ sơ này đang giải quyết ở bước nào sẽ tiếp tục thực hiện theo các bước tương ứng tại quy trình này.
6.1.4. Hồ sơ thay đổi, bổ sung phải trình Hội đồng: căn cứ theo quy định tại điểm a khoản 2 Điều 34 Thông tư số 08/2022/TT-BYT và Chủ trương của Hội đồng đối với việc giải quyết hồ sơ thay đổi, bổ sung trong quá trình lưu hành thuốc, nguyên liệu làm thuốc tại cuộc họp Hội đồng đợt 110.3 thuốc nước ngoài ngày 28/10/2022.
6.1.5. Lãnh đạo Cục, Lãnh đạo phòng, chuyên viên và chuyên gia tham gia thẩm định, xử lý, giải quyết hồ sơ đăng ký thay đổi, bổ sung thuốc sản xuất trong nước, thuốc nhập khẩu, vắc xin, sinh phẩm theo quy trình này phải chịu trách nhiệm trước pháp luật đối với từng công đoạn xử lý hồ sơ. Chuyên viên xử lý hồ sơ phải chủ động, công khai, minh bạch trong quá trình xử lý, trường hợp có khó khăn, vướng mắc cần chủ động xin ý kiến chỉ đạo của Lãnh đạo Phòng, Lãnh đạo Cục để kịp thời giải quyết cho doanh nghiệp.
6.1.6. Thẩm định các tiểu ban: Theo quy định tại Quyết định số 530/QĐ-QLD ngày 28/8/2019 của Cục trưởng Cục Quản lý Dược về việc ban hành Quy chế tổ chức, hoạt động của chuyên gia thẩm định hồ sơ đề nghị cấp, gia hạn, thay đổi, bổ sung giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc.
6.1.7. Thời gian giải quyết trong quy trình này được tính theo ngày làm việc.
6.1.8. Đối với Vắc xin:
- Áp dụng theo quy trình chung như đối với thuốc hóa dược, dược liệu, phóng xạ, sinh phẩm, nguyên liệu làm thuốc.
- Tất cả các bước trong quá trình đăng ký Vắc xin phải được theo dõi, giám sát chặt chẽ và dễ dàng truy xuất theo công cụ phần mềm.
6.2. Quy định về thẩm định hồ sơ thay đổi/bổ sung trình Hội đồng
Thực hiện trình Hội đồng và xử lý sau họp Hội đồng đối với hồ sơ thay đổi/bổ sung thuốc, nguyên liệu làm thuốc sản xuất trong nước, nhập khẩu, vắc xin, sinh phẩm đã được cấp giấy đăng ký lưu hành đối với các trường hợp trình Hội đồng theo quy định tại Điều 34, 35 Thông tư số 08/2022/TT-BYT và chủ trương của Hội đồng đối với hồ sơ thay đổi bổ sung tại Biên bản họp Hội đồng đợt 110.3 thuốc nước ngoài ngày 28/10/2022.
6.3. Quy định về thời gian thẩm định hồ sơ: Quy định tại Điều 38 Thông tư số 08/2022/TT-BYT.
6.4. Nguyên tắc đưa HS ra thẩm định, hoạt động thẩm định HS
- HS hoàn thành thủ tục nộp phí mới được đưa ra thẩm định.
- HS đưa đi thẩm định theo nguyên tắc First In - First Out (FIFO) theo ngày hoàn thành thủ tục nộp phí. HS hoàn thành thủ tục nộp phí trước được đưa ra thẩm định trước, HS hoàn thành thủ tục nộp phí sau được đưa ra thẩm định sau.
- Trường hợp HS cần xử lý ưu tiên để bảo đảm kịp thời nguồn cung ứng thuốc phục vụ điều trị, Phòng có Phiếu trình báo cáo LĐ Cục.
6.5. Quy định về việc công bố công khai đối với các nội dung thay đổi, bổ sung:
6.5.1. Công bố công khai đối với các nội dung thay đổi, bổ sung bao gồm: thông tin ghi trong quyết định cấp giấy đăng ký lưu hành, mẫu nhãn, hướng dẫn sử dụng, thông tin về cơ sở sản xuất nguyên liệu, tiêu chuẩn chất lượng nguyên liệu (áp dụng với hồ sơ sản xuất trong nước, phục vụ việc nhập khẩu nguyên liệu).
6.5.2. Các nội dung thay đổi, bổ sung khác liên quan đến hồ sơ kỹ thuật (trừ nội dung cơ sở sản xuất nguyên liệu, tiêu chuẩn chất lượng nguyên liệu, thông tin tiêu chuẩn chất lượng thuốc thành phẩm):
- Đối với thuốc, nguyên liệu làm thuốc, sinh phẩm: Chỉ được chia sẻ cho các cơ sở đăng ký kèm theo công văn phê duyệt của Cục QLD. Trường hợp nội dung thay đổi bổ sung liên quan đến tiêu chuẩn chất lượng thành phẩm sẽ chia sẻ thêm phần tiêu chuẩn chất lượng (chỉ tiêu và phương pháp kiểm nghiệm) cho các đơn vị kiểm nghiệm thuốc.
- Đối với vắc xin: chỉ được chia sẻ nội dung thay đổi, bổ sung kèm công văn phê duyệt của Cục QLD cho cơ sở đăng ký; chia sẻ nội dung thay đổi, bổ sung kèm công văn phê duyệt và toàn bộ hồ sơ đăng ký thay đổi, bổ sung cho NICVB.
6.5.3. Trường hợp bảng so sánh nội dung đã được phê duyệt và đề nghị thay đổi có cả nội dung được công bố công khai và không được công bố công khai theo quy định nêu tại mục 6.5.1 và 6.5.2 nêu trên, sẽ không công bố công khai bảng so sánh này.
6.6. Nhiệm vụ và trách nhiệm của LĐ Cục, Trưởng phòng ĐKT, LĐP. ĐKT, CVPT, QLHS, CGTĐ/các đơn vị tham gia thẩm định HS:
6.6.1. Lãnh đạo Cục:
6.6.1.1. Cục trưởng:
a) Xem xét số lượng các thuốc thuộc các danh mục hồ sơ trình Hội đồng phù hợp với số lượng các hồ sơ tại Phiếu trình báo cáo của Phòng Đăng ký thuốc, các Phiếu rà soát, biên bản thẩm định hồ sơ và ý kiến của Phó Cục trưởng phụ trách lĩnh vực đăng ký thuốc.
b) Cho ý kiến và sửa đổi, bổ sung (nếu có) liên quan các chủ trương, chính sách dự kiến trình Hội đồng trên cơ sở nội dung đã được rà soát, tổng hợp của Phòng Đăng ký thuốc và ý kiến của Phó Cục trưởng phụ trách lĩnh vực đăng ký thuốc.
c) Xem xét ký hoặc giao Phó Cục trưởng phụ trách lĩnh vực đăng ký thuốc hoặc ủy quyền cho Trưởng phòng Đăng ký thuốc ký công văn chuyển Văn phòng Hội đồng 03 danh mục hồ sơ trình Hội đồng và các chủ trương, chính sách chung trình Hội đồng.
6.6.1.2. Phó Cục trưởng phụ trách lĩnh vực đăng ký thuốc:
a) Xem xét số lượng các thuốc thuộc các danh mục hồ sơ trình Hội đồng phù hợp với số lượng các hồ sơ trình kèm tại Phiếu trình báo cáo của Phòng Đăng ký thuốc, các Phiếu rà soát và biên bản thẩm định hồ sơ.
b) Xem xét việc phân loại thuốc theo đề xuất của Phòng Đăng ký thuốc tại 03 danh mục hồ sơ trình Hội đồng.
c) Xem xét, cho ý kiến đối với đề xuất của Phòng Đăng ký thuốc đối với các nội dung giải trình/ lý do không đồng ý thay đổi, bổ sung các hồ sơ cụ thể cần xin ý kiến Hội đồng.
d) Xem xét, rà soát chung đối với các nội dung chủ trương, chính sách dự kiến trình Hội đồng do Phòng Đăng ký thuốc đề xuất.
đ) Xem xét và ký công văn chuyển Văn phòng Hội đồng danh mục hồ sơ được thẩm định đạt yêu cầu, danh mục hồ sơ được thẩm định không đạt yêu cầu, danh mục hồ sơ đã thẩm định cần xin ý kiến Hội đồng và các chủ trương, chính sách chung trình Hội đồng khi được Cục trưởng phân công.
e) Xem xét và ký phê duyệt công văn đồng ý nội dung thay đổi bổ sung trên cơ sở nội dung đã được P. ĐKT rà soát, tổng hợp.
g) Xem xét và ký quyết định công bố thuốc biệt dược gốc, thuốc có chứng minh tương đương sinh học trên cơ sở nội dung đã được P. ĐKT rà soát, tổng hợp.
6.6.2. Trưởng phòng ĐKT:
Nhiệm vụ và trách nhiệm của TP. ĐKT trong quy trình này bao gồm:
a) Ghi ý kiến đề xuất trên biên bản thẩm định căn cứ ý kiến của Phó Trưởng Phòng phụ trách tại Phiếu rà soát hồ sơ trước khi trình Hội đồng.
b) Rà soát, ký tắt trên từng trang của các danh mục hồ sơ trình Hội đồng sau khi có ý kiến của các Phó Trưởng Phòng phụ trách trên cơ sở xem xét sự phù hợp của danh mục với biên bản thẩm định và các Phiếu rà soát, bao gồm các thông tin sau: Số lượng thuốc, tên thuốc, hoạt chất, dạng bào chế, quy cách đóng gói, tiêu chuẩn, hạn dùng, cơ sở sản xuất, cơ sở đăng ký, tình trạng hồ sơ tương đương sinh học/ hồ sơ lâm sàng/ biệt dược gốc/sinh phẩm tham chiếu (nếu có).
c) Rà soát và ký tắt trên từng trang của nội dung giải trình/ lý do không đồng ý thay đổi, bổ sung các hồ sơ cụ thể cần xin ý kiến Hội đồng sau khi có ý kiến của các Phó Trưởng Phòng được phân công.
d) Rà soát và ký tắt nội dung đề xuất các chủ trương chung trình Hội đồng trên cơ sở tổng hợp của chuyên viên đầu mối tổng hợp chủ trương, chính sách trình Hội đồng và ý kiến của các Phó Trưởng Phòng được phân công.
đ) Ký Phiếu trình báo cáo Lãnh đạo Cục về các danh mục và chủ trương, chính sách trình Hội đồng, trong đó bao gồm số lượng thuốc tại Phiếu trình phù hợp với số lượng tại các danh mục hồ sơ trình Hội đồng.
e) Ký công văn chuyển Văn phòng Hội đồng danh mục hồ sơ và chủ trương chung trình Hội đồng trong trường hợp được Cục trưởng ủy quyền.
g) Chỉ đạo việc xử lý BB họp HĐ, việc triển khai kết luận theo BB họp HĐ và ký tắt các phiếu trình, danh mục.
h) Ký thừa lệnh Cục trưởng công văn thông báo kết quả thẩm định.
i) Rà soát và ký tắt công văn phê duyệt đồng ý thay đổi/bổ sung.
k) Rà soát và ký tắt quyết định công bố thuốc biệt dược gốc, sinh phẩm tham chiếu, thuốc có chứng minh tương đương sinh học.
l) Duyệt kế hoạch điều phối chung trong trường hợp cần thiết để giải quyết hồ sơ tồn đọng.
6.6.3. Phó trưởng Phòng Đăng ký thuốc:
a) Rà soát và ký phê duyệt HS, HS bổ sung đưa ra thẩm định theo nguyên tắc FIFO/ưu tiên.
b) Ghi ý kiến kết luận, ký tên và ghi rõ ngày đề xuất kết luận biên bản. Ý kiến kết luận căn cứ trên kết quả thẩm định của các tiểu ban thẩm định trên BB thẩm định hồ sơ thay đổi/bổ sung, ý kiến của chuyên viên phụ trách và các quy định có liên quan hiện hành.
Nội dung ghi ý kiến kết luận của LĐP trong biên bản thẩm định được ghi cụ thể theo từng loại, bao gồm:
- Hồ sơ đạt/ Hồ sơ xin ý kiến Hội đồng
- Bổ sung hồ sơ
- Không đạt/ Hồ sơ không đạt xin ý kiến Hội đồng
c) Xem xét Phiếu rà soát trình Hội đồng căn cứ các tiêu chí tại Phiếu rà soát, cho ý kiến về nội dung đề xuất, báo cáo của chuyên viên và ký Phiếu rà soát của từng hồ sơ thuộc phạm vi phụ trách.
d) Rà soát, ký tắt trên từng trang của các danh mục hồ sơ trình Hội đồng trên cơ sở xem xét sự phù hợp của danh mục với biên bản thẩm định và các Phiếu rà soát kèm theo, bao gồm các thông tin sau: Số lượng thuốc, tên thuốc, hoạt chất, dạng bào chế, quy cách đóng gói, tiêu chuẩn, hạn dùng, cơ sở sản xuất, cơ sở đăng ký, tình trạng hồ sơ tương đương sinh học/ hồ sơ lâm sàng/ biệt dược gốc/sinh phẩm tham chiếu (nếu có).
đ) Rà soát, ký tắt trên từng trang của nội dung giải trình/ lý do không đồng ý thay đổi, bổ sung cấp các hồ sơ cụ thể cần xin ý kiến Hội đồng.
e) Trường hợp được phân công xem xét các chủ trương, chính sách trình Hội đồng: rà soát và ký tắt nội dung đề xuất các chủ trương chung trình Hội đồng trên cơ sở tổng hợp của chuyên viên đầu mối tổng hợp chủ trương, chính sách trình Hội đồng theo sự phân công của Trưởng Phòng.
g) Đề xuất các chủ trương chung trình HĐ.
h) Ký công văn thông báo kết quả thẩm định hồ sơ theo ủy quyền của Trưởng phòng.
i) Rà soát và ký tắt công văn phê duyệt đồng ý thay đổi/bổ sung theo sự ủy quyền của Trưởng phòng.
k) Rà soát và ký tắt quyết định công bố thuốc biệt dược gốc, sinh phẩm tham chiếu, thuốc có chứng minh tương đương sinh học theo sự ủy quyền của Trưởng phòng.
6.6.4. Chuyên viên phụ trách doanh nghiệp (CVPT): Nhiệm vụ và trách nhiệm của CVPT trong quy trình này bao gồm:
- Rà soát HS lần đầu, HS bổ sung trước khi chuyển thẩm định theo quy định tại Biểu mẫu BM.ĐK.08.08/08.
- Rà soát hồ sơ trước khi giải quyết đối với hồ sơ đã thẩm định xong theo quy định tại Biểu mẫu BM.ĐK.08.08/09.
- Chuyển thẩm định HS theo quy định tại Biểu mẫu BM.ĐK.08.08/12 - Phân loại tiểu ban thẩm định.
- Dự thảo công văn thông báo kết quả thẩm định HS: tổng hợp các ý kiến ghi trong BBTĐ.
- Dự thảo: công văn thông báo kết quả thẩm định HS (tổng hợp các ý kiến ghi trong BBTĐ, ý kiến kết luận của Hội đồng đối với hồ sơ trình Hội đồng); công văn đồng ý phê duyệt thay đổi/bổ sung.
- Lập danh mục thuốc TĐBS trình HĐ đã thẩm định xong để trình HĐ.
- Tổng hợp ký tắt 03 danh mục hồ sơ được thẩm định đạt yêu cầu, danh mục hồ sơ được thẩm định không đạt yêu cầu, danh mục hồ sơ đã thẩm định cần xin ý kiến Hội đồng; cùng với nội dung tổng hợp giải trình/ lý do không đồng ý thay đổi, bổ sung và đề xuất của Phòng Đăng ký thuốc đối với các hồ sơ cụ thể để báo cáo Hội đồng.
- Đối với chuyên viên được phân công: dự thảo nội dung các chủ trương chung trình Hội đồng.
- Lập danh mục hồ sơ để chuyển chuyên viên đầu mối thanh toán cho chuyên gia thẩm định.
- CVPT rà soát các nội dung sau:
+ Hiệu lực giấy đăng ký lưu hành tại thời điểm trình dự thảo công văn.
+ Các thông tin hành chính của thuốc trong đơn đăng ký (bao gồm các thông tin sau: tên thuốc, số đăng ký, hoạt chất, hàm lượng, dạng bào chế, tên và địa chỉ cơ sở sản xuất thành phẩm (nước sản xuất theo đơn đăng ký thay đổi), tên và địa chỉ cơ sở đăng ký) thống nhất theo thông tin đã được Cục Quản lý Dược phê duyệt, theo cơ sở dữ liệu tra cứu của Cục Quản lý Dược phê duyệt ban hành.
- Đối với hồ sơ của thuốc có hoạt chất thuộc danh mục phải thử BE theo quy định không có hồ sơ BE nộp kèm và đơn đăng ký công ty giải trình đã nộp hồ sơ BE trong HS phê duyệt cấp giấy đăng ký lưu hành: CVPT phối hợp với CV đầu mối theo dõi hồ sơ BE của thuốc đăng ký lần đầu kiểm tra, đánh dấu vào nội dung rà soát khi nộp HS BA/BE lần đầu và kèm tài liệu (biên bản thẩm định HS BA/BE đã được phê duyệt cấp số đăng ký) trước khi chuyển chuyên gia các tiểu ban để chuyển thẩm định, bao gồm các thông tin: tên thuốc, số giấy đăng ký lưu hành, cơ sở đăng ký, mã tiếp nhận hồ sơ thay đổi/bổ sung, ngày tiếp nhận hồ sơ thay đổi/bổ sung, thông tin về hồ sơ tương đương sinh học đã nộp lần đầu khi được phê duyệt cấp giấy đăng ký lưu hành.
- Đối với trường hợp hồ sơ Công bố biệt dược gốc/sinh phẩm tham chiếu phải có hồ sơ lâm sàng theo quy định không có HSLS nộp kèm và đơn đăng ký công ty giải trình đã nộp HSLS trong HS phê duyệt cấp giấy đăng ký lưu hành): CVPT phối hợp với CV đầu mối theo dõi HSLS thuốc đăng ký lần đầu tiến hành kiểm tra, đánh dấu vào nội dung rà soát nộp HSLS và kèm tài liệu (biên bản thẩm định HSLS đã được phê duyệt khi cấp số đăng ký lần đầu) trước khi chuyển chuyên gia các tiểu ban để chuyển thẩm định, bao gồm các thông tin: tên thuốc, số giấy đăng ký lưu hành, cơ sở đăng ký, mã tiếp nhận hồ sơ thay đổi/bổ sung, ngày tiếp nhận hồ sơ thay đổi/bổ sung, thông tin về hồ sơ lâm sàng đã nộp lần đầu khi được phê duyệt cấp giấy đăng ký lưu hành.
6.6.5. Chuyên gia thẩm định HS: Nhiệm vụ và trách nhiệm của CGTĐ theo quy chế tổ chức, hoạt động của chuyên gia thẩm định hồ sơ đề nghị cấp, gia hạn, thay đổi, bổ sung giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc còn hiệu lực.
7. NỘI DUNG QUY TRÌNH
7.1. Sơ đồ Quy trình giải quyết hồ sơ thay đổi/bổ sung: (xin xem trang sau)
|
|
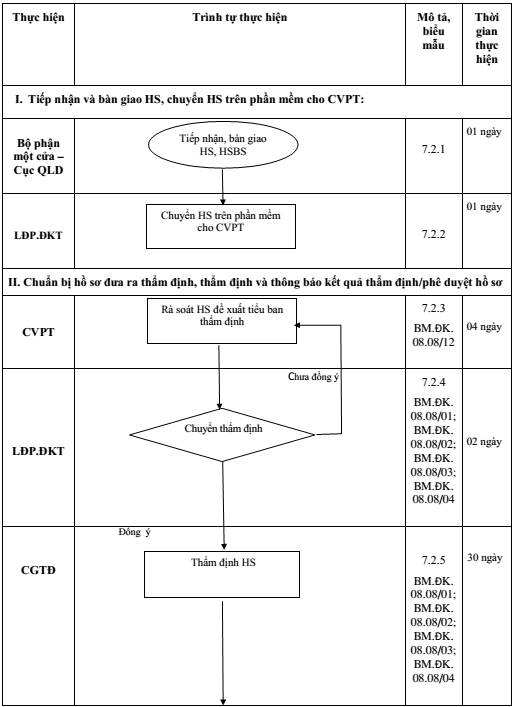
7.2. Mô tả Quy trình giải quyết hồ sơ thay đổi/bổ sung trong quá trình lưu hành
Phần I: Tiếp nhận và bàn giao hồ sơ, chuyển HS trên phần mềm cho CVPT
7.2.1. Tiếp nhận và bàn giao hồ sơ:
Thời gian thực hiện: 01 ngày
Người thực hiện: Bộ phận Một cửa - Cục QLD.
- HS trên hệ thống được chuyển lên Phòng ĐKT là các HS nộp online được xác nhận
đã đóng đủ phí.
* Các lưu ý:
- Trường hợp hồ sơ nộp bản giấy: Doanh nghiệp cập nhật thông tin HS bản giấy hoặc nộp hồ sơ bổ sung (sau khi nhận được công văn online) theo mã HS bản giấy.
- Khi tiếp nhận hồ sơ đề nghị thay đổi bổ sung: Bộ phận Một cửa:
+ Kiểm tra và xác nhận doanh nghiệp nộp đủ phí theo quy định.
+ Kiểm tra thời hạn hiệu lực giấy đăng ký lưu hành để đảm bảo tất cả hồ sơ chuyển lên Phòng Đăng ký thuốc còn hiệu lực giấy ĐKLH tại thời điểm tiếp nhận hồ sơ theo đúng quy định (thời hạn hiệu lực SĐK sẽ tính từ thời điểm DN nộp HS online).
- Đối với trường hợp thay đổi tên cơ sở đăng ký: Cơ sở thực hiện nộp hồ sơ online theo tài khoản của cơ sở đăng ký mới.
- Đối với trường hợp thay đổi cơ sở đăng ký: Cơ sở phải nộp kèm bản scan đơn đăng ký thay đổi, bổ sung có chữ ký xác nhận của cơ sở đăng ký cũ và mới vì đơn đăng ký online chỉ thể hiện chữ ký và dấu xác nhận của một cơ sở. Bộ phận một cửa vẫn thực hiện tiếp nhận hồ sơ bình thường.
7.2.2. Chuyển HS trên phần mềm cho CVPT:
Thời gian thực hiện: 01 ngày
Người thực hiện: Lãnh đạo P. ĐKT.
- HS từ Bộ phận Một cửa - Cục QLD được chuyển lên tài khoản của TP/PTP trong trường hợp được ủy quyền và xuất hiện trong tài khoản của CVPT. Hệ thống sẽ tự động phân công hồ sơ đến tài khoản của CVPT theo bảng phân công nhiệm vụ. Trường hợp, hệ thống không tự động phân công, LĐP thực hiện việc phân công HS vào tài khoản của từng CVPT theo bảng phân công nhiệm vụ. Đối với HSBS: hệ thống sẽ ưu tiên gợi ý CVPT đã thụ lý HS lần đầu.
- Trường hợp HS được chia không đúng CVPT, CVPT chọn trả lại HS để LĐP phân công lại đảm bảo theo đúng Bảng phân công nhiệm vụ. Bên cạnh đó, CVPT có thể chủ động lấy HS theo bảng phân công nhiệm vụ về tài khoản của mình để giải quyết.
Phần II: Chuẩn bị HS đưa ra thẩm định, thẩm định và thông báo kết quả thẩm định/phê duyệt hồ sơ.
7.2.3. Rà soát HS và đề xuất tiểu ban thẩm định
Thời gian thực hiện: 04 ngày
Người thực hiện: CVPT
a) Rà soát HS:
CVPT tiến hành rà soát HS: theo quy định tại Biểu mẫu BM.ĐK.08.08/08.
b) Đề xuất tiểu ban thẩm định:
- Căn cứ vào quy định tại biểu mẫu BM.ĐK.08.08/12
- Phân loại tiểu ban thẩm định, CVPT đề xuất tiểu ban thẩm định HS.
7.2.4. Chuyển HS thẩm định
Thời gian thực hiện: 02 ngày
Người thực hiện: CVPT, LĐP
- Trường hợp đồng ý với đề xuất của CVPT, PTP.ĐKT phân công chuyên gia thẩm định.
- Trường hợp chưa đồng ý với đề xuất của CVPT, chọn trả lại HS cho CVPT, ghi rõ lý do chưa đồng ý hoặc yêu cầu khác.
- Căn cứ duyệt phân loại, rà soát HS: đề xuất của CVPT, tài liệu HS DN kê khai và các tài liệu, quy định có liên quan.
- Nguyên tắc phân công chuyên gia:
+ Căn cứ số lượng HS và tình trạng thực tế của các CG, PTP phụ trách phân bổ hợp lý số lượng HS cho các CG thẩm định.
+ Đối với HS vắc xin, sinh phẩm thẩm định tại Đơn vị thẩm định, PTP chỉ chọn đơn vị, việc phân công CG sẽ do đơn vị thẩm định tự phân công theo quy định của Đơn vị.
- HS trên hệ thống đã được phân công CG sẽ tự động chuyển sang trạng thái đang thẩm định và xuất hiện trên tài khoản của CG.
- Đối với HSBS: hệ thống gợi ý CG thẩm định là các CG đã có tên trên biên bản lần trước. Căn cứ số lượng HS và tình trạng thẩm định HS thực tế của CG, PTP có thể đồng ý với gợi ý của hệ thống hoặc xem xét điều chuyển CG khác.
- Trong trường hợp CG không đảm bảo tiến độ thẩm định hồ sơ theo quy định, PTP phụ trách thu hồi lại HS để điều phối, phân công lại, tránh tồn đọng HS.
* Các Biểu mẫu Biên bản thẩm định:
+ BM.ĐK.08.08/01 và BM.ĐK.08.08/02, BM.ĐK.08.08/02A (đối với hồ sơ không trình Hội đồng).
+ BM.ĐK.08.08/01 và BM.ĐK.08.08/02, BM.ĐK.08.08/02B (đối với hồ sơ trình Hội đồng).
+ BM.ĐK.08.08/01 và BM.ĐK.08.08/02, BM.ĐK.08.08/02B và BM.ĐK.08.08/03 (đối với hồ sơ Công bố biệt dược gốc/sinh phẩm tham chiếu có hồ sơ lâm sàng nộp kèm)
+ BM.ĐK.08.08/01 và BM.ĐK.08.08/02, BM.ĐK.08.08/02B và BM.ĐK.08.08/04 (đối với hồ sơ công bố chứng minh tương đương sinh học có hồ sơ tương đương sinh học nộp kèm).
7.2.5. Thẩm định HS:
Thời gian thực hiện: tối đa 30 ngày.
Người thực hiện: CGTĐ
- CG thẩm định HS theo Quy chế tổ chức, hoạt động của CG thẩm định HS đề nghị cấp, gia hạn, thay đổi, bổ sung giấy ĐKLH thuốc, nguyên liệu làm thuốc.
- Thẩm định trực tuyến, có sẵn mẫu Biên bản, khi CG đánh dấu hết các tiêu chí thẩm định bắt buộc trên Biên bản mới có thể chọn hoàn thành thẩm định.
- Đối với hồ sơ thuốc, nguyên liệu làm thuốc mà tiểu ban thẩm định hồ sơ này có từ 02 chuyên gia trở lên: Trong một tiểu ban thẩm định, CG thẩm định sau có trách nhiệm xem nội dung thẩm định của CG cùng tiểu ban để thống nhất nội dung thẩm định của tiểu ban. Trường hợp có ý kiến khác hoặc phát sinh ý kiến so với CG cùng nhóm thẩm định trước đó, CG thẩm định sau ghi ý kiến vào biên bản để trao đổi với CG thẩm định trước đó hoặc xin ý kiến Cục QLD. Hệ thống cho phép chuyển lại CG một lần để trao đổi, khi đó, HS chuyển lại trạng thái “đang thẩm định”.
- Trường hợp CG thẩm định yêu cầu phân loại lại HS hoặc chuyển nhóm CG hoặc CG khác thẩm định, CG cần ghi rõ vào biên bản để xin ý kiến Cục QLD.
- Mỗi tài khoản CG có đăng ký chữ ký điện tử. Khi CG xác nhận hoàn thành thẩm định, hệ thống sẽ gắn chữ ký điện tử của CG trên biên bản thẩm định. Hệ thống sẽ hiển thị ngày, giờ CG hoàn thành việc thẩm định HS. Khi CG xác nhận hoàn thành việc thẩm định HS, HS được coi là đã thẩm định xong.
- Khi HS đã được thẩm định xong, HS và biên bản thẩm định trên hệ thống được tự động chuyển sang trạng thái đã thẩm định xong tại tài khoản CVPT và LĐP.ĐKT.
- CG thẩm định hồ sơ hoàn toàn trên hệ thống trực tuyến.
- Trong trường hợp CG cần xem hồ sơ bản giấy thì ghi ý kiến vào biên bản thẩm định để Cục QLD có công văn thông báo cơ sở bổ sung bản giấy hồ sơ theo quy định. Khi cơ sở bổ sung hồ sơ bản giấy, CVPT lập danh mục HS chuyển bản giấy theo biểu mẫu BM.ĐK.08.08/13 và trình LĐP phụ trách phê duyệt, chuyển HS kèm danh mục hồ sơ cho CV đầu mối để chuyển CG thẩm định.
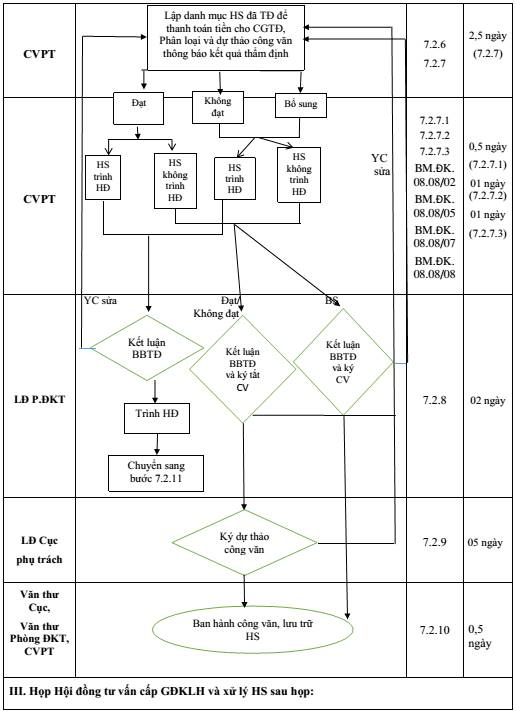
7.2.6. Lập danh mục HS đã TĐ để thanh toán tiền cho CGTĐ:
Thời gian thực hiện: tối đa 10 ngày sau khi CG cuối cùng hoàn thành việc thẩm định. (Thời gian này không được tính trong thời gian giải quyết hồ sơ thay đổi, bổ sung).
Người thực hiện: CVPT, Chuyên viên đầu mối.
CVPT tổng hợp số lượng HS của từng CG của mỗi tiểu ban thẩm định. Vào ngày 30 hàng tháng, CVPT tổng hợp lại toàn bộ số lượng HS đã thẩm định của từng CG trong tháng trên tài khoản của mình để chuyển Chuyên viên đầu mối làm thủ tục thanh toán theo quy định (HS thẩm định lần đầu và HS bổ sung).
7.2.7. Phân loại hồ sơ, tổng hợp ý kiến TĐ và dự thảo công văn thông báo kết quả thẩm định
7.2.7.1. Phân loại hồ sơ:
Thời gian thực hiện tối đa: 0,5 ngày
Người thực hiện: CVPT
Sau khi HS đã được hoàn thành thẩm định, CVPT tổng hợp phân loại HSTĐ/BS trình HĐ và HSTĐ/BS không phải trình HĐ.
- Hồ sơ thay đổi/bổ sung theo quy định tại Phụ lục II ban hành kèm theo Thông tư 08/2022/TT-BYT được phân loại trình Hội đồng theo theo quy định tại điểm a khoản 2 Điều 34 Thông tư 08/2022/TT-BYT và chủ trương Hội đồng đợt 110.3 thuốc nước ngoài khi các tiểu ban thẩm định đạt hoặc đề xuất không đạt, lãnh đạo phòng kết luận “Đạt, trình Hội đồng xin ý kiến” hoặc “Không đạt, trình Hội đồng xin ý kiến”, bao gồm:
+ Thay đổi lớn giấy đăng ký lưu hành thuốc đối với nội dung thay đổi về chỉ định, liều dùng, đối tượng dùng thuốc.
+ Công bố/không công bố biệt dược gốc, sinh phẩm tham chiếu, thuốc có báo cáo nghiên cứu tương đương sinh học.
- HSTĐ/BS không phải trình Hội đồng theo quy định tại điểm a khoản 2 Điều 34 Thông tư 08/2022/TT-BYT và chủ trương Hội đồng đợt 110.3 thuốc nước ngoài.
Lưu ý: Trường hợp không thuộc thay đổi trình Hội đồng, tuy nhiên chuyên gia thẩm định đề xuất xin ý kiến Hội đồng, LĐ.PĐKT căn cứ theo các quy định hiện hành đề xuất kết luận trong BBTĐ “xin ý kiến Hội đồng”.
Đối với HSTĐ/BS không phải trình Hội đồng: tiếp tục thực hiện các bước: 7.2.7.2, 7.2.7.3, 7.2.8, 7.2.9, 7.2.10.
Đối với hồ sơ thay đổi HSTĐ/BS trình Hội đồng: Tiếp tục thực hiện các bước: 7.2.7.2, 7.2.8.1, 7.2.11, 7.2.12, 7.2.13, 7.2.14, 7.2.15.
* Đối với hồ sơ nộp bản giấy:
- Chuyên viên phụ trách lựa chọn mã HS theo Biên bản thẩm định bản giấy, scan Biên bản thẩm định và up vào Hệ thống, thực hiện việc rà soát biên bản sau thẩm định như đối với hồ sơ nộp online.
7.2.7.2. Tổng hợp ý kiến thẩm định:
Thời gian thực hiện: 01 ngày
- Tổng hợp các ý kiến thẩm định của các chuyên gia: đạt/không đạt/bổ sung/ý kiến khác.
- Trường hợp CVPT có ý kiến với ý kiến thẩm định của chuyên gia hoặc bổ sung ý kiến, CVPT ghi ý kiến vào nội dung đề xuất trên BBTĐ - phần xử lý của Phòng ĐKT (mẫu BM.ĐK.08.08/02A hoặc BM.ĐK.08.08/02B) để trình LĐP xem xét.
7.2.7.3. Dự thảo công văn thông báo kết quả thẩm định
Thời gian thực hiện tối đa: 01 ngày
Người thực hiện: CVPT
a) Hồ sơ các tiểu ban thẩm định đề xuất “Đạt”:
- Trên cơ sở ý kiến đề xuất của các tiểu ban, chuyên gia thẩm định hồ sơ đạt yêu cầu, các ý kiến ghi trong BBTĐ, CVPT dự thảo công văn theo mẫu BM.ĐK.08.08/07 và trình Lãnh đạo phòng phụ trách xem xét kết luận biên bản và ký tắt dự thảo công văn để trình Lãnh đạo Cục xem xét.
- Trường hợp CVPT có ý kiến khác với ý kiến đề xuất của Lãnh đạo Phòng, CVPT nêu rõ lý do và ghi cụ thể ý kiến trong BBTĐ, trình lại Lãnh đạo Phòng xem xét kết luận lại, trên cơ sở ý kiến đề xuất của CVPT, LĐP phụ trách xem xét và ghi ý kiến kết luận lại trong BBTĐ. CVPT thực hiện theo ý kiến kết luận ghi trong Biên bản thẩm định.
b) Hồ sơ các tiểu ban thẩm định đề xuất “Bổ sung”:
- Trên cơ sở ý kiến đề xuất của các tiểu ban thẩm định hồ sơ ghi “bổ sung”, các ý kiến ghi trong BBTĐ, CVPT dự thảo công văn theo mẫu BM.ĐK.08.08/05, trình LĐP phụ trách xem xét kết luận biên bản và ký công văn trả lời doanh nghiệp.
b) Hồ sơ các tiểu ban thẩm định đề xuất “Không đạt”:
- Trên cơ sở ý kiến đề xuất của các tiểu ban, CG thẩm định hồ sơ không đạt yêu cầu, các ý kiến ghi trong BBTĐ, CVPT dự thảo công văn theo mẫu BM.ĐK.08.08/06, trình LĐP phụ trách xem xét ghi ý kiến kết luận trên biên bản và ký tắt dự thảo công văn để trình Lãnh đạo Cục xem xét.
7.2.8. Lãnh đạo Phòng xem xét ghi ý kiến biên bản thẩm định và ký tắt dự thảo công văn
Thời gian thực hiện tối đa: 02 ngày
Người thực hiện: LĐ. PĐKT.
7.2.8.1. Ghi ý kiến kết luận BBTĐ
CVPT trình dự thảo công văn, trình cùng hồ sơ kèm theo dự thảo công văn để LĐP xem xét kết luận biên bản và ký thừa ủy quyền Cục trưởng công văn trả lời doanh nghiệp, cụ thể như sau:
- Trên cơ sở các ý kiến chuyên gia thẩm định, ý kiến của chuyên viên, LĐ P.ĐKT ghi kết luận trên BB thẩm định theo 03 hình thức: Thẩm định Bổ sung (Cần bổ sung lại hồ sơ), Thẩm định không đạt (Hồ sơ không đạt yêu cầu), Thẩm định đạt yêu cầu (Hồ sơ đạt yêu cầu), Hồ sơ trình HĐ xin ý kiến.
- Trường hợp có ý kiến với nội dung thẩm định của CGTĐ hoặc bổ sung ý kiến, LĐ P.ĐKT ghi rõ ý kiến, nội dung kết luận, ngày kết luận và ký tên trên BB thẩm định.
- Trường hợp cần trao đổi lại chuyên gia thẩm định, LĐP ghi rõ ý kiến trên BBTĐ, LĐP chuyển BB thẩm định cho CVPT kèm theo ý kiến cần trao đổi để chuyển lại CGTĐ.
* Riêng đối với HS trình HĐ:
- Sau khi LĐP. ĐKT ghi ý kiến kết luận trên BB thẩm định, CVPT tổng hợp danh mục kèm các BB thẩm định trình HĐ (BB có ý kiến kết luận đạt/không đạt/xin ý kiến HĐ) chuyển TP. ĐKT ghi ý kiến đề xuất.
- TP. ĐKT: ghi ý kiến đề xuất vào từng BB thẩm định. Trường hợp có vấn đề liên quan đến chuyên môn, sẽ đề xuất trình xin ý kiến HĐ.
7.2.8.2. Xem xét dự thảo công văn:
a) Trường hợp dự thảo công văn đồng ý hoặc không đồng ý phê duyệt nội dung thay đổi/bổ sung:
Trên cơ sở ý kiến thẩm định của chuyên gia, kết luận tại biên bản thẩm định, LĐP phụ trách xem xét nội dung dự thảo công văn và ký tắt trên bản dự thảo công văn để trình Lãnh đạo Cục xem xét ký công văn đồng ý/không đồng ý phê duyệt.
- Trường hợp yêu cầu sửa, bổ sung công văn, LĐP ghi rõ ý kiến trong BBTĐ và trả lại HS cho CVPT, HS ở trạng thái “LĐP đề nghị rà soát lại”, CVPT thực hiện lại bước 7.2.8.1.
- Trường hợp đề nghị xem lại nội dung thẩm định, LĐP ghi rõ ý kiến trong BBTĐ và chuyển lại CGTĐ, CGTĐ thực hiện lại bước 7.2.5.
- Trường hợp Lãnh đạo Phòng yêu cầu sửa, bổ sung hoặc kiểm tra lại hồ sơ, nội dung thẩm định, LĐP ghi rõ ý kiến trong BBTĐ, CVPT phối hợp với chuyên gia kiểm tra, rà soát lại nội dung thẩm định, báo cáo và trình lại Lãnh đạo Phòng, đề xuất ý kiến giải quyết.
b) Trường hợp dự thảo công văn chưa đồng ý phê duyệt nội dung thay đổi/ bổ sung:
- Dựa trên ý kiến thẩm định của CG, ý kiến của CV, nếu đồng ý với ý kiến CG, ý kiến của CV, LĐP phụ trách xem xét kết luận BBTĐ “Bổ sung” và ký thừa lệnh của Cục trưởng công văn trả lời doanh nghiệp.
- Trường hợp yêu cầu sửa, bổ sung công văn, LĐP ghi rõ ý kiến trong BBTĐ và trả lại HS cho CVPT, HS ở trạng thái “LĐP đề nghị rà soát lại”, CVPT thực hiện lại bước 7.2.8.1.
- Trường hợp đề nghị xem lại nội dung thẩm định, LĐP ghi rõ ý kiến trong BBTĐ và chuyển lại CGTĐ, CGTĐ thực hiện lại bước 7.2.5.
- Trường hợp Lãnh đạo Phòng yêu cầu sửa, bổ sung dự thảo công văn hoặc kiểm tra lại hồ sơ, nội dung thẩm định, LĐP ghi rõ ý kiến trong BBTĐ, CVPT phải báo cáo và trình lại Lãnh đạo Phòng dự thảo công văn, đề xuất ý kiến giải quyết.
- Trong trường hợp, CVPT có ý kiến khác với ý kiến kết luận của Lãnh đạo Phòng, CVPT nêu rõ lý do và ghi cụ thể ý kiến trong BBTĐ, trình lại LĐP xem xét kết luận lại. Trên cơ sở ý kiến đề xuất của CVPT, Lãnh đạo Phòng xem xét và ghi ý kiến kết luận lại trong BBTĐ. CVPT thực hiện theo ý kiến kết luận ghi trong Biên bản thẩm định.
7.2.9. Lãnh đạo Cục xem xét ký công văn
Thời gian thực hiện tối đa: 05 ngày
- Lãnh đạo Cục xem xét và ký công văn trả lời doanh nghiệp.
- Trường hợp Lãnh đạo Cục yêu cầu sửa, bổ sung hoặc kiểm tra lại hồ sơ, nội dung thẩm định và nội dung của công văn, Lãnh đạo Cục ghi rõ ý kiến trong BBTĐ về nội dung có ý kiến, Lãnh đạo Phòng phải báo cáo và trình lại Lãnh đạo Cục nội dung tham mưu, đề xuất giải quyết.
7.2.10. Ban hành công văn
Thời gian thực hiện tối đa: 0,5 ngày
- Văn thư Cục chọn công văn để phát hành, hệ thống sẽ tự động điền số công văn, ngày công văn và gắn dấu của Cục lên văn bản phát hành.
- Văn bản sẽ được trả về tài khoản của doanh nghiệp nộp HS, tài khoản của Viện KNTTW, Viện Kiểm nghiệm thuốc thành phố Hồ Chí Minh (đối với hồ sơ thay đổi, bổ sung về tiêu chuẩn chất lượng thuốc, phương pháp kiểm nghiệm thuốc thành phẩm) theo quy trình.
- Công văn sau khi Lãnh đạo Cục đã ký, hồ sơ và công văn được chuyển cho Văn thư Cục lấy số và đóng dấu và thực hiện phát hành công văn theo quy trình quản lý công văn đi-đến.
- Văn thư Cục trả công văn cho doanh nghiệp đăng ký thuốc (bao gồm cả tài liệu kèm theo, nếu có), chuyển công văn và tài liệu kèm theo cho Viện KNTTW, Viện Kiểm nghiệm thuốc thành phố Hồ Chí Minh (đối với hồ sơ thay đổi, bổ sung về tiêu chuẩn chất lượng thuốc, phương pháp kiểm nghiệm thuốc thành phẩm) theo quy trình.
- Đối với hồ sơ vắc xin: Văn thư Cục trả công văn cho doanh nghiệp đăng ký thuốc (bao gồm cả tài liệu kèm theo, nếu có), chuyển công văn, tài liệu kèm theo (nếu có) và hồ sơ cho NICVB.
Phần III. Họp Hội đồng tư vấn cấp GĐKLH và xử lý HS sau họp:
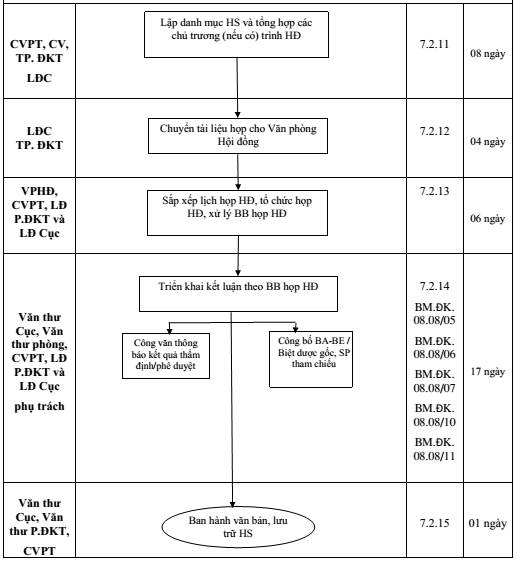
7.2.11. Lập danh mục HS và tổng hợp các chủ trương (nếu có) trình HĐ:
Thời gian thực hiện: 08 ngày.
Người thực hiện: Chuyên viên được phân công, LĐP.ĐKT
* Lập danh mục hồ sơ
- CVPT tổng hợp các danh mục hồ sơ được thẩm định đạt yêu cầu, danh mục hồ sơ được thẩm định không đạt yêu cầu, danh mục hồ sơ đã thẩm định cần xin ý kiến Hội đồng; cùng với nội dung tổng hợp giải trình/ lý do không đồng ý thay đổi, bổ sung và đề xuất của Phòng ĐKT đối với các hồ sơ cụ thể được phân công phụ trách để báo cáo Hội đồng
- PTP ĐKT rà soát, ký tắt trên từng trang của các danh mục hồ sơ trình Hội đồng và rà soát, ký tắt trên từng trang của nội dung giải trình/ lý do không đồng ý thay đổi, bổ sung các hồ sơ cụ thể, trình Trưởng phòng.
- Trưởng phòng ĐKT ký tắt trên từng trang của các danh mục hồ sơ trình Hội đồng, từng trang của nội dung giải trình/ lý do không đồng ý thay đổi, bổ sung, ký Phiếu trình báo cáo Lãnh đạo Cục về các nội dung này.
- Trường hợp được sự phân cấp của Lãnh đạo Cục, các Phó Trưởng phòng phụ trách lĩnh vực trực tiếp rà soát và ký tắt danh mục và Phiếu trình báo cáo trực tiếp Phó Cục trưởng phụ trách lĩnh vực theo mảng công việc được phân công.
* Tổng hợp các chủ trương về ĐKT trình Hội đồng:
- Chuyên viên được phân công dự thảo nội dung các chủ trương chung trình HĐ chuyển PTP ĐKT xem xét.
- Phó Trưởng phòng ĐKT được phân công chịu trách nhiệm rà soát và ký tắt nội dung đề xuất các chủ trương chung trình Hội đồng.
- Trưởng phòng ĐKT ký tắt trên từng trang nội dung đề xuất các chủ trương chung trình Hội đồng và ký Phiếu trình báo cáo Lãnh đạo Cục về các nội dung này.
7.2.12. Chuyển tài liệu họp cho Văn phòng Hội đồng:
Thời gian thực hiện: 04 ngày.
Người thực hiện: Chuyên viên được phân công, LĐP.ĐKT, LĐ Cục QLD
- Phó Cục trưởng phụ trách lĩnh vực đăng ký thuốc xem xét số lượng các thuốc thuộc 03 danh mục hồ sơ trình Hội đồng và xem xét, rà soát chung đối với các nội dung chủ trương, chính sách dự kiến trình Hội đồng.
- Phó Cục trưởng phụ trách lĩnh vực xem xét phân loại các thuốc thuộc 03 danh mục hồ sơ trình Hội đồng; cho ý kiến liên quan các chủ trương, chính sách dự kiến trình Hội đồng.
- Trên cơ sở danh mục hồ sơ và chủ trương chung trình Hội đồng do P. ĐKT đề xuất, Cục trưởng xem xét ký hoặc giao Phó Cục trưởng phụ trách lĩnh vực đăng ký thuốc hoặc ủy quyền Trưởng Phòng ĐKT ký công văn chuyển Văn phòng Hội đồng đối với các nội dung này.
7.2.13. Sắp xếp lịch họp HĐ, tổ chức họp HĐ, xử lý BB họp HĐ
Thời gian thực hiện: 06 ngày.
Người thực hiện: CV được phân công, LĐP. ĐKT, LĐ Cục QLD, VPHĐ
7.2.13.1. Sắp xếp lịch họp HĐ
Thời gian thực hiện: Theo Quy chế của Hội đồng, VPHĐ
Người thực hiện: Hội đồng, VPHĐ
Bước này thực hiện theo Quy chế Tổ chức, hoạt động của Hội đồng tư vấn cấp giấy ĐKLH thuốc, nguyên liệu làm thuốc.
Văn phòng Hội đồng thông báo cho P. ĐKT về lịch họp Hội đồng và các nội dung yêu cầu cần chuẩn bị trước khi tổ chức họp Hội đồng.
7.2.13.2.Tổ chức họp HĐ
Thời gian thực hiện: Theo Quy chế của Hội đồng, VPHĐ
Người thực hiện: Hội đồng, CV, LĐP ĐKT
- Hoạt động của HĐ trước, trong phiên họp HĐ và phối hợp giữa HĐ và Cục QLD sau phiên họp: thực hiện theo Quy chế về tổ chức, hoạt động của Hội đồng tư vấn cấp giấy ĐKLH thuốc, nguyên liệu làm thuốc.
- Mỗi đợt họp Hội đồng có thể được chia làm nhiều phiên họp vào các ngày các nhau; tùy thuộc nội dung họp là các chủ trương trình Hội đồng hoặc tùy thuộc loại và số lượng HS đăng ký thuốc đã hoàn thành thẩm định đạt/ không đạt/ xin ý kiến Hội đồng, đảm bảo phù hợp thời gian họp Hội đồng
7.2.13.3. Xử lý BB họp HĐ:
Văn phòng Hội đồng hoàn thiện Biên bản họp Hội đồng và thông báo cho Cục Quản lý Dược để triển khai các bước tiếp theo: nội dung này thực hiện theo Quy chế về tổ chức, hoạt động của Hội đồng tư vấn cấp giấy ĐKLH thuốc, nguyên liệu làm thuốc.
7.2.14. Triển khai kết luận theo biên bản họp HĐ:
Sau khi nhận được biên bản họp Hội đồng đã có ý kiến kết luận của Chủ tịch Hội đồng từ Văn phòng Hội đồng, Cục Quản lý Dược sẽ tiến hành các bước sau:
a) Hồ sơ TĐ/BS - Hội đồng đồng ý hoặc không đồng ý nội dung thay đổi/ bổ sung:
Thời gian thực hiện: 17 ngày.
- Trên cơ sở kết luận của Hội đồng, CVPT dự thảo công văn theo mẫu BM.ĐK.08.08/06 và BM.ĐK.08.08/07, trình Lãnh đạo phòng phụ trách xem xét ký tắt dự thảo công văn.
- Lãnh đạo Phòng xem xét dự thảo công văn:
+ Trường hợp đồng ý với dự thảo công văn: LĐP ký tắt để chuyển LĐ Cục xem xét, phê duyệt.
+ Trường hợp chưa đồng ý với dự thảo công văn: LĐP chuyển lại cho CVPT thực hiện chỉnh sửa, bổ sung theo nội dung chỉ đạo, ký tắt để trình LĐ Phòng ký tắt để trình LĐ Cục xem xét, phê duyệt.
- Lãnh đạo Cục xem xét dự thảo công văn:
+ Trường hợp đồng ý với dự thảo công văn: LĐC ký phê duyệt công văn.
+ Trường hợp Lãnh đạo Cục yêu cầu sửa, bổ sung nội dung của công văn, Lãnh đạo Cục ghi rõ ý kiến trong công văn dự thảo, Lãnh đạo Phòng phải báo cáo và trình lại Lãnh đạo Cục nội dung tham mưu, đề xuất giải quyết.
b) Hồ sơ TĐ/BS - Hội đồng chưa đồng ý nội dung thay đổi, yêu cầu doanh nghiệp bổ sung:
Thời gian thực hiện: 17 ngày.
- Trên cơ sở kết luận của Hội đồng, CVPT dự thảo công văn theo mẫu
BM.ĐK.08.08/05 và trình Lãnh đạo phòng phụ trách xem xét ký công văn.
- Lãnh đạo Phòng xem xét dự thảo công văn:
+ Trường hợp đồng ý với dự thảo công văn: LĐP xem xét ký công văn.
+ Trường hợp chưa đồng ý với dự thảo công văn: LĐP chuyển lại cho CVPT thực hiện chỉnh sửa, bổ sung theo nội dung chỉ đạo, ký tắt để trình LĐ Phòng ký công văn.
- Sau khi DN nộp hồ sơ bổ sung, giải trình, CVPT thực hiện rà soát và chuyển HS cho CG/TVHĐ thẩm định như sau:
+ Chuyển HSBS lên hệ thống trực tuyến.
+ Trong trường hợp chưa chuyển được HSBS lên hệ thống trực tuyến do hệ thống đang hoàn thiện: CVPT in BBTĐ trên hệ thống trực tuyến, đính kèm BB thẩm định hồ sơ bổ sung theo biểu mẫu BM.ĐK.11.06/17-BS quy định tại Quy trình giải quyết hồ sơ cấp giấy ĐKLH thuốc, nguyên liệu làm thuốc. Bàn giao cho CV đầu mối để chuyển CG/TVHĐ thẩm định.
* Ghi chú: thời gian thực hiện bước này không bao gồm giai đoạn công ty bổ sung sau khi có công văn thông báo ý kiến kết luận của Hội đồng.
c) Đối với hồ sơ Hội đồng đồng ý công bố thuốc thuốc có tài liệu chứng minh tương đương sinh học, thuốc biệt dược gốc, sinh phẩm tham chiếu
Thời gian thực hiện: 17 ngày.
- CVPT căn cứ vào thông tin BB thẩm định để trình Lãnh đạo Phòng phụ trách dự thảo Quyết định công bố danh mục thuốc có tài liệu chứng minh tương đương sinh học (BM.ĐK.08.08/10) hoặc dự thảo Quyết định công bố danh mục thuốc biệt dược gốc/sinh phẩm tham chiếu (BM.ĐK.08.08/11).
- Lãnh đạo phòng phụ trách xem xét dự thảo quyết định:
+ Trường hợp đồng ý với dự thảo Quyết định: LĐP ký tắt dự thảo Quyết định để trình LĐC xem xét, phê duyệt.
+ Trường hợp chưa đồng ý với dự thảo quyết định: LĐP chuyển lại cho CVPT thực hiện chỉnh sửa, bổ sung theo nội dung chỉ đạo, ký tắt để trình LĐ Phòng ký tắt để trình LĐ Cục xem xét, phê duyệt.
- Lãnh đạo Cục xem xét dự thảo quyết định:
+ Trường hợp đồng ý với dự thảo quyết định: Lãnh đạo Cục ký quyết định công bố.
+ Trường hợp Lãnh đạo Cục yêu cầu sửa, bổ sung nội dung của công văn, Lãnh đạo Cục ghi rõ ý kiến trong công văn dự thảo, Lãnh đạo Phòng phải báo cáo và trình lại Lãnh đạo Cục nội dung tham mưu, đề xuất giải quyết.
* Lưu ý: Khi triển khai kết luận theo Biên bản họp Hội đồng, Cục Quản lý Dược phát hiện trường hợp chưa đủ điều kiện để đồng ý phê duyệt khác so với kết luận tại Biên bản họp Hội đồng, Cục Quản lý Dược phải có công văn đề nghị Văn phòng Hội đồng báo cáo Chủ tịch Hội đồng đối với nội dung này. Sau khi có ý kiến kết luận của Chủ tịch Hội đồng, Văn phòng Hội đồng gửi kết luận kèm theo Phiếu trình cho Cục Quản lý Dược để thực hiện các bước tiếp theo.
7.2.15. Ban hành công văn:
Thực hiện như bước 7.2.10.
Thời gian thực hiện: 01 ngày
8. HỒ SƠ:
- Hồ sơ TĐ/BS
- Biên bản thẩm định
9. BIỂU MẪU, PHỤ LỤC:
- BM.ĐK.08.08/01: Mẫu Bìa phân loại hồ sơ thay đổi, bổ sung
- BM.ĐK.08.08/02: Mẫu biên bản thẩm định các nhóm tiểu ban
- BM.ĐK.08.08/02A: Mẫu xử lý của Phòng Đăng ký thuốc (không trình Hội đồng).
- BM.ĐK.08.08/02B: Mẫu xử lý của Phòng Đăng ký thuốc (trình Hội đồng)
- BM.ĐK.08.08/03: Mẫu biên bản thẩm định tiểu ban lâm sàng;
- BM.ĐK.08.08/04: Mẫu biên bản thẩm định tiểu ban BE.
- BM.ĐK. 08.08/05: Mẫu công văn thông báo chưa đồng ý thay đổi, bổ sung;
- BM.ĐK.08.08/06: Mẫu công văn thông báo không đồng ý thay đổi, bổ sung.
- BM.ĐK.08.08/07: Mẫu công văn đồng ý phê duyệt nội dung thay đổi, bổ sung.
- BM.ĐK.08.08/08: Phiếu rà soát hồ sơ trước khi chuyển thẩm định.
- BM.ĐK.08.08/09: Phiếu rà soát hồ sơ trước khi giải quyết đối với hồ sơ đã thẩm định xong.
- BM.ĐK.08.08/10: Quyết định công bố Danh mục thuốc đã được cấp GĐKLH có chứng minh tương đương sinh học.
- BM.ĐK.08.08/11 : Quyết định công bố Danh mục thuốc biệt dược gốc/sinh phẩm tham chiếu.
- BM.ĐK.08.08/12: Phân loại tiểu ban thẩm định hồ sơ thay đổi, bổ sung,
- BM.ĐK.08.08/13: Bàn giao hồ sơ cho chuyên gia thẩm định.
BM.ĐK.08.08/01
Mẫu Bìa Biên bản thẩm định thay đổi/bổ sung thuốc, nguyên liệu làm thuốc đã được cấp số đăng ký lưu hành tại Việt Nam
| BỘ Y TẾ | CỘNG HÒA XÃ HỘI CHỦ NGHĨA VIỆT NAM |
BIÊN BẢN
THẨM ĐỊNH HỒ SƠ ĐĂNG KÝ THAY ĐỔI/ BỔ SUNG THUỐC, NGUYÊN LIỆU LÀM THUỐC ĐÃ ĐƯỢC CẤP GIẤY ĐĂNG KÝ LƯU HÀNH
I. THÔNG TIN CHUNG
1. Tên thuốc:
2. Số đăng ký: Ngày cấp GĐKLH:
3. Hoạt chất:
4. Hàm lượng:
5. Dạng bào chế:
6. Quy cách đóng gói:
7. Hạn dùng:
8. Tiêu chuẩn:
9. Tên công ty sản xuất:
Địa chỉ:
10. Công ty đăng ký:
Địa chỉ:
11. Công văn đến số: Ngày đến:
II. NỘI DUNG ĐĂNG KÝ THAY ĐỔI/BỔ SUNG
1.....
2......
3.....
(Các nội dung khác đề nghị xem tại mục D - Đơn đăng ký thay đổi)
Mẫu BB.08.08/02
| BỘ Y TẾ | CỘNG HÒA XÃ HỘI CHỦ NGHĨA VIỆT NAM |
BIÊN BẢN
THẨM ĐỊNH HỒ SƠ ĐĂNG KÝ THAY ĐỔI/ BỔ SUNG THUỐC, NGUYÊN LIỆU LÀM THUỐC ĐÃ ĐƯỢC CẤP GIẤY ĐĂNG KÝ LƯU HÀNH
| Mã HS lần đầu: | Ngày tiếp nhận: |
1. Ngày đưa hồ sơ ra thẩm định:
2. Tên chuyên viên phụ trách:
3. Phân loại thuốc:
| □ | □ | □ | □ | □ | □ |
| Thuốc hóa dược | Thuốc dược liệu | Vắc xin | Sinh phẩm | Phóng xạ | Nguyên liệu |
4. Nội dung rà soát của chuyên viên phụ trách
Tiểu ban thẩm định hồ sơ
| Tiểu ban pháp chế | Tiểu ban chất lượng | Tiểu ban bào chế | Tiểu ban dược lý | Tiểu ban lâm sàng | Tiểu ban BA/BE |
| □ | □ | □ | □ | □ | □ |
| Loại hồ sơ | Trong nước | □ | Nước ngoài | □ |
| Thuốc hóa dược | Thuốc dược liệu | Thuốc hóa dược | Thuốc dược liệu | |
| □ | □ | □ | □ |
Mẫu BB.08.08/02
PHẦN A. NỘI DUNG VÀ KẾT QUẢ THẨM ĐỊNH
(Ngày đưa hồ sơ thẩm định: / /20…….)
Ý KIẾN THẨM ĐỊNH TIỂU BAN PHÁP CHẾ
1. NỘI DUNG THẨM ĐỊNH (Đề nghị tích vào ô: Đạt/ Bổ sung /Không đạt/Không yêu cầu)
| Tên mục | Nội dung | Ý kiến thẩm định | |||
| Đạt | Bổ sung | Không đạt | Không yêu cầu | ||
| 1 | Đơn đăng ký: thông tin thống nhất với các phần trong hồ sơ, phân loại nội dung thay đổi/bổ sung phù hợp với các quy định hiện hành |
|
|
|
|
| 2 | Điều kiện cần đáp ứng đối với từng nội dung đề nghị thay đổi quy định tại Phụ lục II Thông tư số 32/2018/TT-BYT |
|
|
|
|
| 3 | Hồ sơ cần nộp đối với từng nội dung đề nghị thay đổi quy định tại Phụ lục II Thông tư số 32/2018/TT-BYT |
|
|
|
|
| 2. Ý KIẾN CHI TIẾT CỦA CHUYÊN GIA …….
|
| Ngày thẩm định | Họ và tên chuyên gia | Chữ ký | Đề xuất của Tiểu ban…. |
|
| Đạt □ Bổ sung □ Không đạt □ | ||||
|
|
|
|
| |
|
|
|
|
|
Thẩm định hồ sơ bổ sung - Tiểu ban Pháp chế
Hồ sơ đăng ký thay đổi/bổ sung
Hồ sơ bổ sung lần 1:
Hồ sơ bổ sung lần 2:
Hồ sơ bổ sung lần 3:
| Thẩm định hồ sơ bổ sung | Họ và tên chuyên gia | Chữ ký | Đề xuất của Tiểu ban pháp chế | |||
| Lần | Ngày thẩm định | Đạt | Bổ sung | Không đạt | ||
| 1 |
|
|
|
|
|
|
| 2 |
|
|
|
|
|
|
Ý KIẾN THẨM ĐỊNH TIỂU BAN TIÊU CHUẨN CHẤT LƯỢNG
1. NỘI DUNG THẨM ĐỊNH (Đề nghị tích vào ô Đạt/ Bổ sung /Không đạt/Không yêu cầu)
| Tên mục | Nội dung | Ý kiến thẩm định | |||
| Đạt | Bổ sung | Không đạt | Không yêu cầu | ||
| 1 | Nội dung đề nghị thay đổi/bổ sung trong Đơn đăng ký: thông tin thống nhất với các phần trong hồ sơ |
|
|
|
|
| 2 | Điều kiện cần đáp ứng đối với từng nội dung đề nghị thay đổi quy định tại Phụ lục II Thông tư số 32/2018/TT-BYT |
|
|
|
|
| 3 | Hồ sơ cần nộp đối với từng nội dung đề nghị thay đổi quy định tại Phụ lục II Thông tư số 32/2018/TT-BYT |
|
|
|
|
| 2. Ý KIẾN CHI TIẾT CỦA CHUYÊN GIA CHẤT LƯỢNG
|
| Ngày thẩm định | Họ và tên chuyên gia | Chữ ký | Đề xuất của Tiểu ban chất lượng |
|
| Đạt □ Bổ sung □ Không đạt □ | ||||
|
|
|
|
| |
|
|
|
|
|
Thẩm định hồ sơ bổ sung - Tiểu ban chất lượng
Hồ sơ đăng ký thay đổi/bổ sung
Hồ sơ bổ sung lần 1:
Hồ sơ bổ sung lần 2:
Hồ sơ bổ sung lần 3:
| Thẩm định hồ sơ bổ sung | Họ và tên chuyên gia | Chữ ký | Đề xuất của Tiểu ban chất lượng | |||
| Lần | Ngày thẩm định | Đạt | Bổ sung | Không đạt | ||
| 1 |
|
|
|
|
|
|
| 2 |
|
|
|
|
|
|
Ý KIẾN TH ẨM ĐỊNH CỦA TIỂU BAN BÀO CHẾ
1. NỘI DUNG THẨM ĐỊNH (Đề nghị tích vào ô Đạt/ Bổ sung /Không đạt/Không yêu cầu)
| Tên mục | Nội dung | Ý kiến thẩm định | |||
| Đạt | Bổ sung | Không đạt | Không yêu cầu | ||
| 1 | Nội dung đề nghị thay đổi/bổ sung trong Đơn đăng ký: thông tin thống nhất với các phần trong hồ sơ |
|
|
|
|
| 2 | Điều kiện cần đáp ứng đối với từng nội dung đề nghị thay đổi quy định tại Phụ lục II Thông tư số 32/2018/TT-BYT |
|
|
|
|
| 3 | Hồ sơ cần nộp đối với từng nội dung đề nghị thay đổi quy định tại Phụ lục II Thông tư số 32/2018/TT-BYT |
|
|
|
|
| 2. Ý KIẾN CHI TIẾT CỦA CHUYÊN GIA BÀO CHẾ
|
| Ngày thẩm định | Họ và tên chuyên gia | Chữ ký | Đề xuất của Tiểu ban bào chế |
|
| Đạt □ Bổ sung □ Không đạt □ | ||||
|
|
|
|
| |
|
|
|
|
|
Thẩm định hồ sơ bổ sung - Tiểu ban bào chế
Hồ sơ đăng ký thay đổi/bổ sung
Hồ sơ bổ sung lần 1:
Hồ sơ bổ sung lần 2:
Hồ sơ bổ sung lần 3:
| Thẩm định hồ sơ bổ sung | Họ và tên chuyên gia | Chữ ký | Đề xuất của Tiểu ban bào chế | |||
| Lần | Ngày thẩm định | Đạt | Bổ sung | Không đạt | ||
| 1 |
|
|
|
|
|
|
| 2 |
|
|
|
|
|
|
Ý KIẾN THẨM ĐỊNH CỦA TIỂU BAN DƯỢC LÝ
1. NỘI DUNG THẨM ĐỊNH (Đề nghị tích vào ô: Đạt/ Bổ sung /Không đạt/Không yêu cầu)
| Tên mục | Nội dung | Ý kiến thẩm định | |||
| Đạt | Bổ sung | Không đạt | Không yêu cầu | ||
| 1 | Nội dung đề nghị thay đổi/bổ sung trong Đơn đăng ký: thông tin thống nhất với các phần trong hồ sơ |
|
|
|
|
| 2 | Điều kiện cần đáp ứng đối với từng nội dung đề nghị thay đổi quy định tại Phụ lục II Thông tư số 32/2018/TT-BYT |
|
|
|
|
| 3 | Hồ sơ cần nộp đối với từng nội dung đề nghị thay đổi quy định tại Phụ lục II Thông tư số 32/2018/TT-BYT |
|
|
|
|
| 4 | Tờ hướng dẫn sử dụng dự kiến thay đổi |
|
|
|
|
| 2. Ý KIẾN CHI TIẾT CỦA CHUYÊN GIA DƯỢC LÝ
|
| Ngày thẩm định | Họ và tên chuyên gia | Chữ ký | Đề xuất của Tiểu ban dược lý |
|
| Đạt □ Bổ sung □ Không đạt □ | ||||
|
|
|
|
| |
|
|
|
|
|
Thẩm định hồ sơ bổ sung - Tiểu ban dược lý
Hồ sơ đăng ký thay đổi/bổ sung
Hồ sơ bổ sung lần 1:
Hồ sơ bổ sung lần 2:
Hồ sơ bổ sung lần 3:
| Thẩm định hồ sơ bổ sung | Họ và tên chuyên gia | Chữ ký | Đề xuất của Tiểu ban dược lý | |||
| Lần | Ngày thẩm định | Đạt | Bổ sung | Không đạt | ||
| 1 |
|
|
|
|
|
|
| 2 |
|
|
|
|
|
|
Ý KIẾN CỦA CHUYÊN GIA THẨM ĐỊNH TIỂU BAN LÂM SÀNG (trường hợp không có hồ sơ lâm sàng nhưng có các tài liệu cần xin thêm ý kiến tiểu ban lâm sàng hoặc trường hợp trình Hội đồng đối với bổ sung chỉ định, liều dùng, đối tượng dùng)
1. NỘI DUNG THẨM ĐỊNH (Đề nghị tích vào ô: Đạt/ Bổ sung /Không đạt/Không yêu cầu)
| Tên mục | Nội dung | Ý kiến thẩm định | |||
| Đạt | Bổ sung | Không đạt | Không yêu cầu | ||
| 1 | Nội dung đề nghị thay đổi/bổ sung trong Đơn đăng ký: thông tin thống nhất với các phần trong hồ sơ |
|
|
|
|
| 2 | Điều kiện cần đáp ứng đối với từng nội dung đề nghị thay đổi quy định tại Phụ lục II Thông tư số 32/2018/TT-BYT |
|
|
|
|
| 3 | Hồ sơ cần nộp đối với từng nội dung đề nghị thay đổi quy định tại Phụ lục II Thông tư số 32/2018/TT-BYT |
|
|
|
|
| 4 | Tờ hướng dẫn sử dụng dự kiến thay đổi |
|
|
|
|
| 2. Ý KIẾN CHI TIẾT CỦA CHUYÊN GIA LÂM SÀNG
|
| Ngày thẩm định | Họ và tên chuyên gia | Chữ ký | Đề xuất của Tiểu ban lâm sàng |
|
| Đạt □ Bổ sung □ Không đạt □ | ||||
|
|
|
|
| |
|
|
|
|
|
Thẩm định hồ sơ bổ sung - Tiểu ban lâm sàng
Hồ sơ đăng ký thay đổi/bổ sung
Hồ sơ bổ sung lần 1:
Hồ sơ bổ sung lần 2:
Hồ sơ bổ sung lần 3:
| Thẩm định hồ sơ bổ sung | Họ và tên chuyên gia | Chữ ký | Đề xuất của Tiểu ban lâm sàng | |||
| Lần | Ngày thẩm định | Đạt | Bổ sung | Không đạt | ||
| 1 |
|
|
|
|
|
|
| 2 |
|
|
|
|
|
|
| 3 |
|
|
|
|
|
|
Ý KIẾN CỦA CHUYÊN GIA THẨM ĐỊNH TIỂU BAN BA/BE (trường hợp không có hồ sơ BABE nhưng có các tài liệu cần xin thêm ý kiến tiểu ban BA/BE)
1. NỘI DUNG THẨM ĐỊNH ((Đề nghị tích vào ô: Đạt/Bổ sung/Không đạt/Không yêu cầu)
| Tên mục | Nội dung | Ý kiến thẩm định | |||
| Đạt | Bổ sung | Không đạt | Không yêu cầu | ||
| 1 | Nội dung đề nghị thay đổi/bổ sung trong Đơn đăng ký: thông tin thống nhất với các phần trong hồ sơ |
|
|
|
|
| 2 | Điều kiện cần đáp ứng đối với từng nội dung đề nghị thay đổi quy định tại Phụ lục II Thông tư số 32/2018/TT-BYT |
|
|
|
|
| 3 | Hồ sơ cần nộp đối với từng nội dung đề nghị thay đổi quy định tại Phụ lục II Thông tư số 32/2018/TT-BYT |
|
|
|
|
| 2. Ý KIẾN CHI TIẾT CỦA CHUYÊN GIA BA/BE
|
| Ngày thẩm định | Họ và tên chuyên gia | Chữ ký | Đề xuất của Tiểu ban BA/BE |
|
|
|
| Đạt □ Bổ sung □ Không đạt □ |
|
|
|
| |
|
|
|
|
Thẩm định hồ sơ bổ sung - Tiểu ban BA/BE
Hồ sơ đăng ký thay đổi/bổ sung
Hồ sơ bổ sung lần 1:
Hồ sơ bổ sung lần 2:
Hồ sơ bổ sung lần 3:
| Thẩm định hồ sơ bổ sung | Họ và tên chuyên gia | Chữ ký | Đề xuất của Tiểu ban BA/BE | |||
| Lần | Ngày thẩm định | Đạt | Bổ sung | Không đạt | ||
| 1 |
|
|
|
|
|
|
| 2 |
|
|
|
|
|
|
| 3 |
|
|
|
|
|
|
BM.ĐK.08.08/03: Mẫu Biên bản thẩm định nhóm Lâm sàng
Ý KIẾN CỦA CHUYÊN GIA THẨM ĐỊNH TIỂU BAN LÂM SÀNG
1. CÁC NỘI DUNG THẨM ĐỊNH (Đề nghị tích vào ô: Đạt/Không yêu cầu/Chưa đạt/Không đạt)
| Tên mục | Nội dung | Ý kiến thẩm định | |||
| Đạt | Bổ sung | Không đạt | Không yêu cầu | ||
| 1. | Nhóm dược lý (ghi rõ) |
|
|
|
|
| I. | Trường hợp không có hồ sơ lâm sàng |
|
|
|
|
|
| Thuốc đã nộp hồ sơ lâm sàng và được thẩm định hồ sơ lâm sàng trước khi cấp số đăng ký lưu hành đối với hồ sơ theo ACTD hoặc ICH-CTD (đơn đăng ký) |
|
|
|
|
|
| Tờ hướng dẫn sử dụng dự kiến thay đổi |
|
|
|
|
|
| Các tài liệu lâm sàng tham khảo nộp kèm |
|
|
|
|
|
| Các nội dung khác liên quan (Biên bản thẩm định hồ sơ lâm sàng đã thẩm định do phòng Đăng ký thuốc cung cấp kèm theo) |
|
|
|
|
| II | Trường hợp có hồ sơ lâm sàng |
|
|
|
|
| A. | Phần A: Tổng quan lâm sàng | ||||
| 2. | Cơ sở phát triển sản phẩm |
|
|
|
|
| 3. | Tổng quan về sinh dược học |
|
|
|
|
| 4. | Tổng quan về dược lý lâm sàng |
|
|
|
|
| 5. | Tổng quan về hiệu quả |
|
|
|
|
| 6. | Tổng quan về an toàn |
|
|
|
|
| 7. | Kết luận về lợi ích và nguy cơ |
|
|
|
|
| B. | Phần B: Tóm tắt lâm sàng | ||||
| 8. | Tóm tắt các nghiên cứu về sinh dược học và phương pháp phân tích 8.1. Cơ sở nghiên cứu và tổng quan 8.2. Tóm tắt kết quả các nghiên cứu riêng lẻ 8.3. So sánh và phân tích kết quả xuyên suốt các nghiên cứu. |
|
|
|
|
| 9. | Tóm tắt các nghiên cứu về dược lý lâm sàng 9.1. Cơ sở nghiên cứu và tổng quan 9.2. Tóm tắt kết quả các nghiên cứu riêng lẻ 9.3. So sánh và phân tích kết quả xuyên suốt các nghiên cứu 9.4. Các nghiên cứu đặc biệt. |
|
|
|
|
| 10. | Tóm tắt về hiệu quả lâm sàng 10.1. Cơ sở nghiên cứu và tổng quan về hiệu quả lâm sàng 10.2. Tóm tắt kết quả các nghiên cứu riêng lẻ 10.3. So sánh và phân tích kết quả xuyên suốt các nghiên cứu 10.4. Phân tích các thông tin lâm sàng liên quan đến các khuyến cáo về liều dùng 10.5. Sự duy trì hiệu quả và/hoặc sự nhờn thuốc |
|
|
|
|
| 11. | Tóm tắt về tính an toàn lâm sàng 11.1. Mức độ sử dụng thuốc 11.2. Biến cố ngoại ý 11.3. Đánh giá kết quả xét nghiệm 11.4. Dấu hiệu sinh tồn, triệu chứng thực thể và các ghi nhận khác liên quan đến tính an toàn 11.5. Sự an toàn đối với các nhóm bệnh nhân đặc biệt và các tình huống đặc biệt |
|
|
|
|
| 12. | Bản tóm tắt các nghiên cứu riêng lẻ |
|
|
|
|
| C. | Phần C: Bảng danh sách tất cả các nghiên cứu lâm sàng |
|
|
|
|
| D. | Phần D: Báo cáo nghiên cứu lâm sàng | ||||
| 13. | Báo cáo các nghiên cứu sinh dược học 13.1. Báo cáo nghiên cứu sinh khả dụng (BA) 13.2. Báo cáo nghiên cứu so sánh sinh khả dụng hoặc tương đương sinh học (BE) 13.3. Báo cáo nghiên cứu tương quan in vitro - in vivo 13.4. Báo cáo các phương pháp phân tích sinh học và phương pháp phân tích sử dụng cho các nghiên cứu ở người |
|
|
|
|
| 14. | Báo cáo các nghiên cứu liên quan đến dược động học (PK) sử dụng nguyên liệu sinh học từ người 14.1. Báo cáo nghiên cứu sự gắn kết với protein huyết tương 14.2. Báo cáo nghiên cứu sự chuyển hóa ở gan và tương tác thuốc 14.3. Báo cáo các nghiên cứu sử dụng nguyên liệu sinh học từ người khác |
|
|
|
|
| E. | Phần E: Báo cáo nghiên cứu lâm sàng | ||||
| 15. | Báo cáo các nghiên cứu về dược động học (PK) trên người 15.1. Báo cáo nghiên cứu về PK và sự dung nạp ban đầu trên người khỏe mạnh 15.2. Báo cáo nghiên cứu về PK và sự dung nạp ban đầu trên người bệnh 15.3. Báo cáo nghiên cứu PK trên dân số |
|
|
|
|
| 16. | Báo cáo các nghiên cứu về dược lực học (PD) trên người 16.1. Báo cáo nghiên cứu về PK và PK/PD trên người khỏe mạnh 16.2. Báo cáo nghiên cứu về PK và PK/PD trên người bệnh |
|
|
|
|
| 17. | Báo cáo các nghiên cứu về hiệu quả và tính an toàn 17.1. Báo cáo các nghiên cứu lâm sàng có đối chứng liên quan đến chỉ định đề nghị 17.2. Báo cáo các nghiên cứu lâm sàng không có đối chứng 17.3. Báo cáo phân tích các dữ liệu từ nhiều nghiên cứu, bao gồm tất cả các phân tích tích hợp (integrated analysis), phân tích gộp (meta-analysis) và phân tích bắc cầu (bridging analysis) chính thức 17.4. Các báo cáo nghiên cứu lâm sàng khác |
|
|
|
|
| 18. | Báo cáo các kinh nghiệm sau khi đưa thuốc ra thị trường |
|
|
|
|
| 19. | Mẫu báo cáo dữ liệu và danh sách các người bệnh |
|
|
|
|
| F. | Phần F: Danh mục các tài liệu tham khảo chủ yếu |
|
|
|
|
| G. | Phần G: Nội dung tờ hướng dẫn sử dụng | ||||
| 20. | Hoạt chất quy đổi ra dạng tính liều điều trị (không bắt buộc đối với vắc xin, sinh phẩm) |
|
|
|
|
| 21. | Chỉ định |
|
|
|
|
| 22. | Cách dùng, liều dùng |
|
|
|
|
| 23. | Đặc tính dược lực học (không bắt buộc đối với thuốc không kê đơn) |
|
|
|
|
| 24. | Đặc tính dược động học (không bắt buộc đối với vắc xin, thuốc không kê đơn) |
|
|
|
|
| 25. | Chống chỉ định |
|
|
|
|
| 26. | Cảnh báo và thận trọng khi dùng thuốc |
|
|
|
|
| 27. | Tá dược cần bổ sung cảnh báo thận trọng |
|
|
|
|
| 28. | Sử dụng thuốc cho phụ nữ có thai và cho con bú |
|
|
|
|
| 29. | Ảnh hưởng của thuốc lên khả năng lái xe, vận hành máy móc |
|
|
|
|
| 30. | Tương kỵ, tương tác thuốc |
|
|
|
|
| 31. | Tác dụng không mong muốn của thuốc |
|
|
|
|
| 32. | Quá liều và cách xử trí |
|
|
|
|
| 33. | Các nội dung khác (nếu có) |
|
|
|
|
| 2. Ý KIẾN CHI TIẾT CỦA CHUYÊN GIA LÂM SÀNG Mục 1: ..... (Ý kiến thẩm định cụ thể) ….
|
| Ngày thẩm định | Tên chuyên gia | Chữ ký | Đề xuất của tiểu ban lâm sàng |
|
|
|
| Đạt □ Chưa đạt □ Không đạt □ |
|
|
|
| |
|
|
|
|
Thẩm định hồ sơ bổ sung tiểu ban Lâm sàng
Hồ sơ đăng ký thay đổi/bổ sung
Hồ sơ bổ sung lần 1:
Hồ sơ bổ sung lần 2:
Hồ sơ bổ sung lần 3:
| Thẩm định hồ sơ bổ sung | Họ và tên chuyên gia | Chữ ký | Đề xuất của Tiểu ban Lâm sàng | |||
| Lần | Ngày thẩm định | Đạt | Bổ sung | Không đạt | ||
| 1 |
|
|
|
|
|
|
| 2 |
|
|
|
|
|
|
| 3 |
|
|
|
|
|
|
BM.ĐK.08.08/04
Ý KIẾN CỦA CHUYÊN GIA THẨM ĐỊNH TIỂU BAN TƯƠNG ĐƯƠNG SINH HỌC
2. CÁC NỘI DUNG THẨM ĐỊNH (Đề nghị ghi rõ: Đạt/Không yêu cầu/Chưa đạt/Không đạt)
| Tên mục | Nội dung | Ý kiến thẩm định | |||||||
| Đạt | Bổ sung | Không đạt | Không yêu cầu | ||||||
| I | Trường hợp không nộp hồ sơ BA/BE: | ||||||||
| 1. | Các tài liệu nộp kèm hồ sơ liên quan hồ sơ BA/BE |
|
|
|
| ||||
| 2. | Các nội dung khác kèm theo (Biên bản thẩm định HS BA/BE do phòng Đăng ký thuốc cung cấp kèm theo) |
|
|
|
| ||||
| II | Trường hợp nộp kèm hồ sơ BA/BE |
|
|
|
| ||||
| A | Cơ sở nghiên cứu và xác nhận năng lực: | ||||||||
| 1. | Cơ sở nghiên cứu (Tên và địa chỉ: cơ sở lâm sàng; cơ sở phân tích; bộ phận phân tích và quản lý dữ liệu) |
|
|
|
| ||||
| 2. | Trang chữ ký: - Tên, chữ ký và ngày ký của nghiên cứu viên chính, phụ trách lâm sàng, phụ trách phân tích, phụ trách phân tích thống kê - Danh sách các nghiên cứu viên khác tham gia nghiên cứu |
|
|
|
| ||||
| 3. | Xác nhận năng lực cơ sở nghiên cứu (Giấy chứng nhận/ chứng chỉ GCP và GLP/ biên bản thanh tra) |
|
|
|
| ||||
| B | Đề cương nghiên cứu : (kiểm tra sự phù hợp của thiết kế nghiên cứu với thuốc nghiên cứu theo các quy định hiện hành) |
|
|
|
| ||||
| 4. | Thiết kế nghiên cứu (chéo/song song/lặp lại): |
|
|
|
| ||||
| 5. | Thuyết minh về cỡ mẫu: ghi rõ Số người tình nguyện: |
|
|
|
| ||||
| 6. | Kiểu nghiên cứu (đói/ no - đơn liều/ đa liều): |
|
|
|
| ||||
| 7. | Chất phân tích: |
|
|
|
| ||||
| 8. | Ý kiến khác (Mù hóa, Ngẫu nhiên hóa, Tiêu chuẩn hóa nghiên cứu): |
|
|
|
| ||||
| C | Bản chấp thuận của Hội đồng đạo đức: | ||||||||
| 9. | Số đề cương: |
|
|
|
| ||||
| 10. | Ngày họp hội đồng/ ngày phê duyệt: |
|
|
|
| ||||
| D | Thuốc dùng trong nghiên cứu: | ||||||||
|
| Thuốc thử | ||||||||
| 11. | - Kiểm tra sự phù hợp của thuốc thử dùng trong nghiên cứu với thuốc đăng ký (hoạt chất, hàm lượng, dạng bào chế, tên nhà sản xuất, địa điểm sản xuất) |
|
|
|
| ||||
| 12. | - Phiếu kiểm nghiệm |
|
|
|
| ||||
| 13. | - Cỡ lô SX |
|
|
|
| ||||
|
| Thuốc đối chứng | ||||||||
| 14. | - Tên thương mại, hoạt chất, hàm lượng, dạng bào chế, tên nhà sản xuất, địa điểm sản xuất (yêu cầu ghi cụ thể, rõ ràng): |
|
|
|
| ||||
| 15. | - Đáp ứng chí lựa chọn thuốc đối chứng ? |
|
|
|
| ||||
| 16. | - Thuốc đối chứng dùng trong nghiên cứu có tại Việt Nam? |
|
|
|
| ||||
| 17. | Trường hợp thuốc chứng đã dùng trong nghiên cứu khác với thuốc đối chứng có tại Việt Nam: + Nguồn gốc, xuất xứ thuốc đối chứng: + Tương đương độ hòa tan giữa thuốc đối chứng dùng trong nghiên cứu và thuốc đối chứng có tại Việt Nam (lưu ý: có thể chấp nhận khác lô) |
|
|
|
| ||||
| 18. | Tương đương về chất lượng giữa thuốc thử và thuốc đối chứng (tương đương độ hòa tan, chênh lệch hàm lượng): |
|
|
|
| ||||
| E | Thực nghiệm và kết quả nghiên cứu: (Kiểm tra sự phù hợp với đề cương nghiên cứu đã được phê duyệt) | ||||||||
|
| Kết quả nghiên cứu lâm sàng | ||||||||
| 19. | - Người tình nguyện |
|
|
|
| ||||
| 20. | - Thời gian thực hiện các pha lâm sàng: từng giai đoạn và thời gian nghỉ giữa các giai đoạn. |
|
|
|
| ||||
| 21. | - Dùng thuốc: + Bảng ngẫu nhiên hóa và trình tự dùng thuốc của người tình nguyện; + Các ý kiến khác |
|
|
|
| ||||
| 22. | - Lấy mẫu |
|
|
|
| ||||
| 23. | - Phiếu chấp thuận tình nguyện tham gia nghiên cứu (copy) |
|
|
|
| ||||
| 24. | - Dữ liệu báo cáo lâm sàng: Báo cáo sai lệch so với đề cương, biến cố bất lợi và hồ sơ của người tình nguyện có biến cố bất lợi (nếu có), quá trình thực hiện theo quy định. |
|
|
|
| ||||
|
| Thẩm định quy trình phân tích | ||||||||
| 25. | - Chất phân tích (Dược chất gốc/ Chất chuyển hóa), phương pháp phân tích hoặc điều kiện phân tích |
|
|
|
| ||||
| 26. | - Kết quả thẩm định: Tính đặc hiệu, Độ đúng, Độ chính xác, Độ phục hồi, Tính tuyến tính, Giới hạn định lượng dưới; Độ nhiễm chéo và ảnh hưởng của nền mẫu (với pp LC/MS) |
|
|
|
| ||||
| 27. | - Độ ổn định (của các dung dịch gốc và mẫu sinh học trong các điều kiện xử lý mẫu và bảo quản) |
|
|
|
| ||||
| 28. | - Sắc ký đồ thẩm định đại diện (tính đặc hiệu, tính tuyến tính, độ ổn định dài ngày, độ nhiễm chéo và ảnh hưởng của nền mẫu (với phương pháp LC/MS) |
|
|
|
| ||||
|
| Kết quả phân tích mẫu người tình nguyện: | ||||||||
| 29. | - Ngày tiến hành/ kết thúc (*). |
|
|
|
| ||||
| 30. | - Bảng tóm tắt quá trình phân tích, mã hóa mẫu, sắc đồ, kết quả đánh giá sự phù hợp của lô phân tích và thông tin về phân tích lại. |
|
|
|
| ||||
| 31. | - Bảng kết quả nồng độ chất phân tích trong huyết tương của thuốc thử/ thuốc chứng thu được từ từng cá thể |
|
|
|
| ||||
| 32. | - 20% sắc ký đồ của người tình nguyện |
|
|
|
| ||||
|
| Báo cáo dược động học | ||||||||
| 33. | - Bảng các thông số dược động học cơ bản và dữ liệu thống kê: Cmax, Tmax, AUC0-t và AUC0-int của thuốc thử và thuốc chứng trên từng cá thể |
|
|
|
| ||||
| 34. | - Đường biểu diễn nồng độ thuốc - thời gian trung bình và của từng người tình nguyện |
|
|
|
| ||||
| 35. | - Bảng tỷ số các thông số Cmax; AUC0-t và AUC0-int đã chuyển logarit của thử/ chứng trên từng cá thể |
|
|
|
| ||||
| 36. | - Bảng thông số dược động học khác: T1/2, số điểm tính Kel, tỷ số AUCt/AUCint và các thông số khác (nếu có) |
|
|
|
| ||||
|
| Phân tích thống kê | ||||||||
| 37. | - Phân tích phương sai các giá trị Cmax, AUC0-t và AUC0-int (có giá trị Power) và các thông số khác (nếu có) |
|
|
|
| ||||
| 38. | - So sánh Tmax theo phương pháp thống kê phi tham số |
|
|
|
| ||||
| 39. | - Kết quả khoảng tin cậy 90% của tỷ số Cmax; AUC0-t và AUC0-int trên số liệu đã chuyển logarit |
|
|
|
| ||||
| F | Hồ sơ đề nghị miễn thử TĐSH in vivo: | ||||||||
| 40. | Bảng so sánh công thức, tỷ lệ hàm lượng các thành phần dược chất, tá dược. |
|
|
|
| ||||
| 41. | Bảng so sánh QTSX (Tóm tắt): |
|
|
|
| ||||
| 42. | Dược động học tuyến tính: |
|
|
|
| ||||
| 43. | Báo cáo kết quả thử nghiệm tương đương độ hòa tan |
|
|
|
| ||||
| 44. | Báo cáo thẩm định quy trình phân tích xác định lượng dược chất hòa tan |
|
|
|
| ||||
| G | 7. Bàn luận (nếu có) và Kết luận (tương đương sinh học/ không tương đương sinh học)
| ||||||||
| 2. Ý KIẾN CHI TIẾT CỦA CHUYÊN GIA TƯƠNG ĐƯƠNG SINH HỌC Mục 1: ..... (Ý kiến thẩm định cụ thể) ....
|
| Ngày thẩm định | Họ và tên chuyên gia | Chữ ký | Đề xuất của Tiểu ban BA/BE |
|
|
|
| Đạt □ Bổ sung □ Không đạt □ |
|
|
|
| |
|
|
|
|
Thẩm định hồ sơ bổ sung tiểu ban BA/BE
Hồ sơ đăng ký thay đổi/bổ sung
Hồ sơ bổ sung lần 1:
Hồ sơ bổ sung lần 2:
Hồ sơ bổ sung lần 3:
| Thẩm định hồ sơ bổ sung | Họ và tên chuyên gia | Chữ ký | Đề xuất của Tiểu ban BA/BE | |||
| Lần | Ngày thẩm định | Đạt | Bổ sung | Không đạt | ||
| 1 |
|
|
|
|
|
|
| 2 |
|
|
|
|
|
|
| 3 |
|
|
|
|
|
|
BM.ĐK.08.08/05
Mẫu công văn thông báo chưa đồng ý thay đổi, bổ sung
| BỘ Y TẾ | CỘNG HÒA XÃ HỘI CHỦ NGHĨA VIỆT NAM |
| Số: /QLD-ĐK | Hà Nội, ngày tháng năm 20… |
| Kính gửi: |
|
Cục Quản lý Dược nhận được hồ sơ số [số hồ sơ tiếp nhận] ngày [ngày tiếp nhận] và các tài liệu liên quan của công ty về việc thay đổi, bổ sung đối với thuốc ......., số đăng ký ..........;
Căn cứ Thông tư số 08/2022/TT-BYT ngày 05/9/2022 của Bộ Y tế quy định việc đăng ký lưu hành thuốc, nguyên liệu làm thuốc;
Căn cứ Biên bản thẩm định hồ sơ thay đổi/bổ sung của công ty, Cục Quản lý Dược thông báo kết quả thẩm định hồ sơ như sau:
- ....
- ....
- ....
Sau 12 tháng kể từ ngày ký công văn này, công ty không nộp tài liệu bổ sung thì hồ sơ đã nộp không còn giá trị. (áp dụng đối với các trường hợp bổ sung tài liệu khác)
Sau 36 tháng kể từ ngày ký công văn này, công ty không nộp tài liệu bổ sung thì hồ sơ đã nộp không còn giá trị. (áp dụng đối với các trường hợp bổ sung tài liệu lâm sàng, tiền lâm sàng, tài liệu tương đương sinh học, tài liệu nghiên cứu độ ổn định)
Cục Quản lý Dược thông báo để công ty biết và thực hiện đúng các quy định về đăng ký lưu hành thuốc./.
|
| TL. CỤC TRƯỞNG |
BM.ĐK.08.08/06
Mẫu công văn thông báo không đồng ý thay đổi, bổ sung
| BỘ Y TẾ | CỘNG HÒA XÃ HỘI CHỦ NGHĨA VIỆT NAM |
| Số: /QLD-ĐK | Hà Nội, ngày tháng năm 20… |
| Kính gửi: |
|
Cục Quản lý Dược nhận được hồ sơ số [số hồ sơ tiếp nhận] ngày [ngày tiếp nhận] và các tài liệu liên quan của công ty về việc thay đổi, bổ sung đối với thuốc đã được cấp giấy đăng ký lưu hành;
Căn cứ Thông tư số 08/2022/TT-BYT ngày 05/9/2022 của Bộ Y tế quy định việc đăng ký lưu hành thuốc, nguyên liệu làm thuốc;
Căn cứ Biên bản thẩm định hồ sơ thay đổi/bổ sung của công ty, Cục Quản lý Dược có ý kiến như sau:
Không đồng ý với nội dung đề nghị thay đổi/bổ sung đối với thuốc ......., số đăng ký .........., lý do:
- ....
- ....
- ....
Cục Quản lý Dược thông báo để công ty biết và thực hiện đúng các quy định về đăng ký lưu hành thuốc./.
|
| KT. CỤC TRƯỞNG |
BM.ĐK.08.08/07
Mẫu công văn phê duyệt nội dung thay đổi, bổ sung
BM.ĐK.08.08/07A
Mẫu công văn phê duyệt nội dung thay đổi, bổ sung (Trả lời hồ sơ không trình Hội đồng)
| BỘ Y TẾ | CỘNG HÒA XÃ HỘI CHỦ NGHĨA VIỆT NAM |
| Số: /QLD-ĐK | Hà Nội, ngày tháng năm 20… |
| Kính gửi: |
|
Trả lời hồ sơ số tiếp nhận số [mã hồ sơ doanh nghiệp nộp] ngày [ngày nộp hs] và các tài liệu liên quan của công ty về việc thay đổi, bổ sung đối với thuốc đã được cấp giấy đăng ký lưu hành;
Căn cứ Thông tư số 08/2022/TT-BYT ngày 05/9/2022 của Bộ Y tế quy định việc đăng ký lưu hành thuốc, nguyên liệu làm thuốc;
Căn cứ Biên bản thẩm định hồ sơ thay đổi/bổ sung của công ty, Cục Quản lý Dược có ý kiến như sau:
Đồng ý với nội dung đề nghị thay đổi, bổ sung được phê duyệt kèm theo công văn này đối với thuốc [tên thuốc], số đăng ký [số đăng ký].
Ngoài nội dung được phê duyệt, các nội dung khác giữ nguyên như hồ sơ đăng ký thuốc lưu tại Cục Quản lý Dược.
Các nội dung thay đổi, bổ sung nêu trên được thực hiện kể từ ngày ký công văn này. Riêng nội dung thay đổi, sau 12 tháng kể từ ngày ký công văn này, công ty phải thực hiện theo nội dung thay đổi đã được phê duyệt.
Cục Quản lý Dược thông báo để công ty biết và thực hiện đúng các quy định về đăng ký lưu hành thuốc./.
|
| KT. CỤC TRƯỞNG |
BM.ĐK.08.08/07B
Mẫu công văn phê duyệt nội dung thay đổi, bổ sung (Trả lời hồ sơ trình Hội đồng)
| BỘ Y TẾ | CỘNG HÒA XÃ HỘI CHỦ NGHĨA VIỆT NAM |
| Số: /QLD-ĐK | Hà Nội, ngày tháng năm 20… |
| Kính gửi: |
|
Trả lời hồ sơ số tiếp nhận số [mã hồ sơ doanh nghiệp nộp] ngày [ngày nộp hs] và các tài liệu liên quan của công ty về việc thay đổi, bổ sung đối với thuốc đã được cấp giấy đăng ký lưu hành;
Căn cứ Thông tư số 08/2022/TT-BYT ngày 05/9/2022 của Bộ Y tế quy định việc đăng ký lưu hành thuốc, nguyên liệu làm thuốc;
Căn cứ Biên bản họp Hội đồng tư vấn cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc, Cục Quản lý Dược có ý kiến như sau:
Đồng ý với nội dung đề nghị thay đổi, bổ sung được phê duyệt kèm theo công văn này đối với thuốc [tên thuốc], số đăng ký [số đăng ký].
Ngoài nội dung được phê duyệt, các nội dung khác giữ nguyên như hồ sơ đăng ký thuốc lưu tại Cục Quản lý Dược.
Các nội dung thay đổi, bổ sung nêu trên được thực hiện kể từ ngày ký công văn này. Riêng nội dung thay đổi, sau 12 tháng kể từ ngày ký công văn này, công ty phải thực hiện theo nội dung thay đổi đã được phê duyệt.
Cục Quản lý Dược thông báo để công ty biết và thực hiện đúng các quy định về đăng ký lưu hành thuốc./.
|
| KT. CỤC TRƯỞNG |
BM.ĐK. 08.08/08
PHIẾU RÀ SOÁT BIÊN BẢN THẨM ĐỊNH TRƯỚC KHI CHUYỂN THẨM ĐỊNH
| Stt | Nội dung rà soát | Đạt | Không đạt |
|
| Thuốc không có trong quyết định thu hồi đã được Cục Quản lý Dược ban hành |
|
|
| Ngày tháng năm | Ngày tháng năm |
BM.ĐK.08.08/09
PHIẾU RÀ SOÁT HỒ SƠ TRƯỚC KHI GIẢI QUYẾT ĐỐI VỚI HỒ SƠ ĐÃ THẨM ĐỊNH XONG
| Stt | Nội dung rà soát | Đạt | Không đạt |
| 1 | 'Hồ sơ đã được sửa đổi, bổ sung không quá 03 lần (1) |
|
|
| 2 | 'Biên bản đầy đủ chữ ký, ngày thẩm định của chuyên gia thẩm định (2) |
|
|
| 3 | Hồ sơ bổ sung đã được chuyển các tiểu ban liên quan thẩm định bao gồm: |
|
|
| 3.1 | - Các nội dung bổ sung phát sinh từ yêu cầu của tiểu ban thẩm định này nhưng có liên quan đến nội dung phải thẩm định của tiểu ban thẩm định khác |
|
|
| 3.2 | - Các nội dung thay đổi/ bổ sung phát sinh theo đề nghị của doanh nghiệp trong công văn đề nghị liên quan tiểu ban thẩm định đạt trước đó |
|
|
|
| Đề xuất: Hồ sơ notify đạt, đề nghị công bố Hồ sơ TĐ/BS thẩm định đạt Hồ sơ TĐ/BS thẩm định đạt, trình HĐ Hồ sơ thẩm định không đạt Hồ sơ bổ sung Đề xuất nhóm chuyên gia để thẩm định | ||
| Ngày tháng năm | Ngày tháng năm |
* Ghi chú:
(1): Áp dụng đối với hồ sơ nộp theo quy định tại Thông tư 08/2022/TT-BYT;
(2): Áp dụng đối với biên bản thẩm định giấy;
BM.ĐK.08.08/10
| BỘ Y TẾ | CỘNG HÒA XÃ HỘI CHỦ NGHĨA VIỆT NAM |
| Số: /QĐ-QLD | Hà Nội, ngày tháng năm |
QUYẾT ĐỊNH
VỀ VIỆC CÔNG BỐ DANH MỤC THUỐC CÓ TÀI LIỆU CHỨNG MINH TƯƠNG ĐƯƠNG SINH HỌC - ĐỢT ...
CỤC QUẢN LÝ DƯỢC
Căn cứ Luật Dược số 105/2016/QH13 ngày 06/04/2016;
Căn cứ Nghị định 54/2017/NĐ-CP ngày 08/05/2017 của Chính phủ quy định chi tiết một số điều về biện pháp thi hành Luật Dược;
Căn cứ Nghị định số 155/2018/NĐ-CP ngày 12/11/2018 của Chính phủ sửa đổi một số quy định liên quan đến điều kiện đầu tư kinh doanh thuộc phạm vi quản lý nhà nước của Bộ Y tế;
Căn cứ Nghị định số 75/2017/NĐ-CP ngày 20/6/2017 của Chính phủ quy định chức năng, nhiệm vụ, quyền hạn và cơ cấu tổ chức của Bộ Y tế;
Căn cứ Thông tư số 08/20228/TT-BYT ngày 05/9/2022 của Bộ trưởng Bộ Y tế quy định việc đăng ký lưu hành thuốc, nguyên liệu làm thuốc;
Căn cứ Quyết định số 1969/QĐ-BYT ngày 26/4/2023 của Bộ trưởng Bộ Y tế quy định chức năng, nhiệm vụ, quyền hạn và cơ cấu tổ chức của Cục Quản lý Dược thuộc Bộ Y tế;
Căn cứ kết luận của Hội đồng tư vấn cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc ... đợt ... họp ngày...;
Xét đề nghị của Trưởng phòng Phòng Đăng ký thuốc - Cục Quản lý Dược,
QUYẾT ĐỊNH:
Điều 1. Công bố kèm theo Quyết định này Danh mục thuốc có tài liệu chứng minh tương đương sinh học - Đợt... gồm ...thuốc.
Điều 2. Quyết định này có hiệu lực kể từ ngày ký ban hành.
Điều 3. Giám đốc Sở Y tế các tỉnh, thành phố trực thuộc Trung ương, các cơ sở kinh doanh dược và Giám đốc cơ sở có thuốc nêu tại Điều 1 chịu trách nhiệm thi hành Quyết định này./.
|
| CỤC TRƯỞNG |
DANH MỤC
THUỐC CÓ TÀI LIỆU CHỨNG MINH TƯƠNG ĐƯƠNG SINH HỌC (ĐỢT...)
(Ban hành kèm theo Quyết định số .../QĐ-QLD ngày ../.../.... của Cục trưởng Cục Quản lý Dược)
| STT | Tên thuốc | Hoạt chất | Hàm lượng | Dạng bào chế | Quy cách đóng gói | Số đăng ký | Cơ sở sản xuất | Địa chỉ cơ sở sản xuất | Nước sản xuất |
| ... |
|
|
|
|
|
|
|
|
|
BM.ĐK.08.08/11
| BỘ Y TẾ | CỘNG HÒA XÃ HỘI CHỦ NGHĨA VIỆT NAM |
| Số: /QĐ-QLD | Hà Nội, ngày tháng năm |
QUYẾT ĐỊNH
VỀ VIỆC CÔNG BỐ DANH MỤC THUỐC BIỆT DƯỢC GỐC/SINH PHẨM THAM CHIẾU - ĐỢT ...
CỤC QUẢN LÝ DƯỢC
Căn cứ Luật Dược số 105/2016/QH13 ngày 06/04/2016;
Căn cứ Nghị định 54/2017/NĐ-CP ngày 08/05/2017 của Chính phủ quy định chi tiết một số điều về biện pháp thi hành Luật Dược;
Căn cứ Nghị định số 155/2018/NĐ-CP ngày 12/11/2018 của Chính phủ sửa đổi một số quy định liên quan đến điều kiện đầu tư kinh doanh thuộc phạm vi quản lý nhà nước của Bộ Y tế;
Căn cứ Nghị định số 75/2017/NĐ-CP ngày 20/6/2017 của Chính phủ quy định chức năng, nhiệm vụ, quyền hạn và cơ cấu tổ chức của Bộ Y tế;
Căn cứ Thông tư số 08/2022/TT-BYT ngày 05/9/2022 của Bộ trưởng Bộ Y tế quy định việc đăng ký lưu hành thuốc, nguyên liệu làm thuốc;
Căn cứ Quyết định số 1969/QĐ-BYT ngày 26/4/2023 của Bộ trưởng Bộ Y tế quy định chức năng, nhiệm vụ, quyền hạn và cơ cấu tổ chức của Cục Quản lý Dược thuộc Bộ Y tế;
Căn cứ kết luận của Hội đồng tư vấn cấp giấy đăng ký lưu hành thuốc, nguyên liệu làm thuốc ... đợt ... họp ngày...;
Xét đề nghị của Trưởng phòng Phòng Đăng ký thuốc - Cục Quản lý Dược,
QUYẾT ĐỊNH:
Điều 1. Công bố kèm theo Quyết định này Danh mục thuốc biệt dược gốc/sinh phẩm tham chiếu - Đợt... gồm ...thuốc.
Điều 2. Quyết định này có hiệu lực kể từ ngày ký ban hành.
Điều 3. Giám đốc Sở Y tế các tỉnh, thành phố trực thuộc Trung ương, các cơ sở kinh doanh dược và Giám đốc cơ sở có thuốc nêu tại Điều 1 chịu trách nhiệm thi hành Quyết định này./.
|
| CỤC TRƯỞNG |
DANH MỤC
THUỐC BIỆT DƯỢC GỐC/SINH PHẨM THAM CHIẾU (ĐỢT...)
(Ban hành kèm theo Quyết định số .../QĐ-QLD ngày ../.../.... của Cục trưởng Cục Quản lý Dược)
| STT | Tên thuốc/sinh phẩm | Hoạt chất | Hàm lượng | Dạng bào chế | Quy cách đóng gói | Số đăng ký | Cơ sở sản xuất | Địa chỉ cơ sở sản xuất | Nước sản xuất |
| ... |
|
|
|
|
|
|
|
|
|
BM.ĐK.08.08/12
PHÂN LOẠI TIỂU BAN THẨM ĐỊNH HỒ SƠ THAY ĐỔI, BỔ SUNG
A. THUỐC HÓA DƯỢC
THAY ĐỔI LỚN (MaV)
| Nội dung thay đổi | Pháp chế | Tiêu chuẩn | Bào chế | Dược lý | Lâm sàng | BA/BE | |
| Nội dung thay đổi 1 (MaV- 1) | Thay đổi và/hoặc bổ sung chỉ định/liều dùng/đối tượng bệnh nhân/bổ sung thông tin lâm sàng để mở rộng phạm vi sử dụng thuốc | x |
|
| x | x |
|
| Điều kiện cần đáp ứng (C) | 1. Là kết quả của thay đổi Tóm tắt đặc tính sản phẩm (SmPC) hoặc tài liệu tương đương (như USPI) dẫn đến thay đổi hướng dẫn sử dụng và/hoặc mẫu nhãn 2. Không áp dụng đối với trường hợp các thuốc generic khi cập nhật tờ hướng dẫn sử dụng theo tờ hướng dẫn sử dụng của biệt dược gốc tại Việt Nam hoặc theo tờ hướng dẫn sử dụng đã được các cơ quan quản lý SRA phê duyệt của thuốc không có biệt dược gốc tại Việt Nam. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Hướng dẫn sử dụng và/hoặc mẫu nhãn đã được duyệt. 2. Hướng dẫn sử dụng và/hoặc mẫu nhãn mới (02 bản). Bảng so sánh các nội dung thay đổi của hướng dẫn sử dụng (02 bản) và/hoặc mẫu nhãn. Trường hợp nộp hồ sơ trực tuyến chỉ yêu cầu nộp 01 bản hướng dẫn sử dụng và/hoặc mẫu nhãn mới, 01 bản bảng so sánh. 3. Giải trình lý do thay đổi. 4. Tài liệu tham khảo và/hoặc Báo cáo của các chuyên gia lâm sàng (nếu có). 5. Hướng dẫn sử dụng/Tóm tắt đặc tính sản phẩm /Thông tin cho bệnh nhân được chấp thuận bởi cơ quan cấp phép lưu hành thuốc của nước sở tại hoặc nước tham chiếu cho phép thay đổi hoặc công văn của cơ quan cấp phép lưu hành thuốc của nước sở tại hoặc nước tham chiếu cho phép thay đổi/bổ sung (đối với thuốc sản xuất tại nước ngoài có thay đổi dựa trên thay đổi tại nước sở tại hoặc nước tham chiếu) 6. Tài liệu lâm sàng theo Hồ sơ kỹ thuật chung ASEAN (ACTD) phần IV |
|
|
|
|
|
|
| Nội dung thay đổi 2 (MaV-2) | Thay đổi/bổ sung nội dung của hướng dẫn sử dụng và/hoặc mẫu nhãn | x |
|
| x | x |
|
| Điều kiện cần đáp ứng (C) | 1. Thay đổi này không phải là thay đổi nhỏ và không nằm trong phạm vi MaV-1. 2. Là kết quả của thay đổi Tóm tắt đặc tính sản phẩm (SmPC) hoặc tài liệu tương đương (như USPI). |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Hướng dẫn sử dụng và/hoặc mẫu nhãn đã được duyệt. 2. Hướng dẫn sử dụng và/hoặc mẫu nhãn mới. Bảng so sánh các nội dung thay đổi của hướng dẫn sử dụng và/hoặc mẫu nhãn. 3. Giải trình lý do thay đổi. 4. Tài liệu tham khảo và/hoặc các tài liệu lâm sàng chứng minh. 5. Hướng dẫn sử dụng (PI)/ Tóm tắt đặc tính sản phẩm (SmPC)/Thông tin cho bệnh nhân (PIL) được chấp thuận bởi cơ quan cấp phép lưu hành thuốc của nước sở tại hoặc nước tham chiếu cho phép thay đổi/bổ sung (đối với thuốc sản xuất tại nước ngoài có thay đổi dựa trên thay đổi tại nước sở tại hoặc nước tham chiếu) |
|
|
|
|
|
|
| Nội dung thay đổi 3 (MaV-3) | Thay đổi và/hoặc bổ sung cơ sở sản xuất/địa điểm sản xuất dược chất (khi không có Giấy chứng nhận tuân thủ Dược điển châu Âu (CEP)) |
|
|
|
|
|
|
| TH1 | Các thuốc có chứng minh TĐSH | x | x | x |
|
| x |
| TH2 | Các thuốc không có chứng minh TĐSH | x | x | x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Tiêu chuẩn chất lượng dược chất không thay đổi. 2. Trường hợp thay đổi và/hoặc bổ sung cơ sở sản xuất/địa điểm sản xuất của dược chất khi có Giấy chứng nhận tuân thủ Dược điển châu Âu (CEP) áp dụng MiV-PA4. 3. Trường hợp có thay đổi tiêu chuẩn chất lượng dược chất, áp dụng thêm quy định tại MAV-6 hoặc MiV- PA8 hoặc MiV-N6. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Có thể nộp một trong những tài liệu sau: a) Toàn bộ các phần từ S1-S7 theo ACTD; b) Hồ sơ tổng thể hoạt chất bao gồm cả phần công khai và phần không công khai (phần không công khai được cơ sở sản xuất dược chất cung cấp trực tiếp cho Cục Quản lý Dược kèm theo thư cho phép tiếp cận tài liệu này); c) Giấy chứng nhận hoặc tài liệu kiểm tra tương đương được cấp từ một trong các nước tham chiếu quy định tại Thông tư đăng ký thuốc. 2. Bảng so sánh sự khác nhau trong quy trình sản xuất dược chất tại địa điểm sản xuất mới và địa điểm sản xuất đã được duyệt (nếu có thay đổi). 3. Phiếu kiểm nghiệm và/hoặc số liệu phân tích lô (dưới dạng bảng so sánh) của cơ sở sản xuất dược chất trên ít nhất 02 lô pilot giữa dược chất sản xuất tại địa điểm mới và dược chất sản xuất tại địa điểm đã được duyệt. Đối với trường hợp thuốc được công bố có chứng minh tương đương sinh học, bảng so sánh phân tích lô phải bao gồm cả các chỉ tiêu chất lượng có trong tiêu chuẩn chất lượng đã được duyệt của dược chất và các chỉ tiêu chất lượng không có trong tiêu chuẩn nhưng đã được thiết lập trong quá trình nghiên cứu phát triển thành phẩm (là các chỉ tiêu đã được mô tả tại phần P2. Phát triển dược học trong hồ sơ đăng ký thuốc, ví dụ: kích thước tiểu phân, dạng thù hình, trạng thái hydrat hoá/solvat hoá, đặc tính hòa tan, khối lượng riêng biểu kiến, độ trơn chảy của dược chất) kèm theo tài liệu chứng minh nếu cần. 4. Số liệu so sánh biểu đồ hòa tan giữa các thuốc thành phẩm trước và sau khi thay đổi đối với trường hợp thuốc đã được công bố có tài liệu chứng minh tương đương sinh học. 5. Thư cam kết của cơ sở sản xuất thuốc thành phẩm về việc tiến hành nghiên cứu độ ổn định của thuốc thành phẩm được sản xuất từ dược chất sản xuất tại địa điểm mới ở điều kiện dài hạn và điều kiện lão hóa cấp tốc và sẽ báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành, đồng thời đề xuất biện pháp xử lý phù hợp. 6. Giấy tờ pháp lý của cơ sở sản xuất dược chất chứng minh cơ sở đáp ứng thực hành tốt sản xuất nguyên liệu làm thuốc theo quy định tại khoản 11 Điều 22 Thông tư này. Trường hợp nguyên liệu đã có Giấy đăng ký lưu hành tại Việt Nam, không yêu cầu tài liệu quy định tại khoản 1, 5 mục này. |
|
|
|
|
|
|
| Nội dung thay đổi 4 (MaV-4) | Thay đổi địa điểm sản xuất: a) bán thành phẩm ở dạng bulk product; b) thuốc thành phẩm. | x | x | x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Không áp dụng cho những thay đổi liên quan đến cơ sở chịu trách nhiệm xuất xưởng lô hoặc địa điểm chỉ xuất xưởng lô. 2. Cơ sở sản xuất thuốc thành phẩm/ bán thành phẩm ở dạng bulk product không thay đổi. 3. Địa điểm sản xuất mới phải đạt GMP theo quy định phù hợp với dạng bào chế của thuốc. Đối với thuốc sản xuất tại Việt Nam, địa điểm sản xuất mới phải đã được cấp Giấy chứng nhận đủ điều kiện kinh doanh thuốc. 4. Thay đổi cơ sở/ địa điểm cơ sở chịu trách nhiệm xuất xưởng lô áp dụng MiV-PA3. 5. Nếu có thay đổi về quy trình sản xuất, áp dụng thêm MaV-9. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Đối với thuốc sản xuất tại nước ngoài: CPP hoặc giấy chứng nhận GMP chứng minh địa điểm sản xuất mới phù hợp để được sản xuất dạng bào chế của thuốc. 2. Bảng so sánh số liệu phân tích lô giữa ít nhất 02 lô sản xuất (hoặc 01 lô sản xuất và 02 lô pilot) của thuốc thành phẩm/ bán thành phẩm ở dạng bulk product sản xuất tại địa điểm mới và 03 lô sản xuất cuối cùng của thuốc thành phẩm/ bán thành phẩm ở dạng bulk product sản xuất tại địa điểm đã được duyệt. Số liệu phân tích lô cho 02 lô sản xuất tiếp theo của thuốc thành phẩm/ bán thành phẩm ở dạng bulk product tại địa điểm mới phải sẵn có để cung cấp khi có yêu cầu. Trong quá trình phân tích lô, nếu có kết quả nào không đạt tiêu chuẩn chất lượng phải có báo cáo đồng thời đề xuất biện pháp xử lý phù hợp. 3. Số liệu độ ổn định của thuốc thành phẩm sau khi thay đổi và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 4. Kế hoạch thẩm định và/hoặc báo cáo thẩm định quy trình sản xuất thuốc thành phẩm/ bán thành phẩm ở dạng bulk product tại địa điểm mới. 5. Đối với các thuốc có dạng bào chế rắn dùng đường uống: Số liệu so sánh biểu đồ hòa tan giữa các thuốc thành phẩm/ bán thành phẩm ở dạng bulk product trước và sau khi có thay đổi. 6. Công thức bào chế đã được duyệt của thuốc thành phẩm. 7. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. 8. Tiêu chuẩn chất lượng đã được duyệt của dược chất. 9. Nghiên cứu ảnh hưởng của thời gian lưu trữ bán thành phẩm ở dạng bulk product trong quá trình bảo quản và vận chuyển từ nơi sản xuất bán thành phẩm đến nơi đóng gói sơ cấp. 10. Đối với cơ sở sản xuất bán thành phẩm ở dạng bulk product theo hợp đồng, thư chỉ định và cho phép sản xuất thuốc bán thành phẩm ở dạng bulk product tại địa điểm mới trong đó nêu rõ các công đoạn nào của quy trình sản xuất được thực hiện tại địa điểm này. |
|
|
|
|
|
|
| Nội dung thay đổi 5 (MaV-5) | Thay đổi địa điểm cơ sở đóng gói sơ cấp | x |
| x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Không có sự thay đổi nào ngoại trừ thay đổi địa điểm của cơ sở đóng gói sơ cấp. 2. Địa điểm đóng gói mới phải đạt GMP theo quy định phù hợp với dạng bào chế của thuốc. Đối với thuốc đóng gói tại Việt Nam, địa điểm đóng gói mới phải đã được cấp Giấy chứng nhận đủ điều kiện kinh doanh thuốc. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Đối với thuốc đóng gói tại nước ngoài: CPP hoặc giấy chứng nhận GMP chứng minh địa điểm đóng gói mới phù hợp để được đóng gói sơ cấp dạng bào chế của thuốc. 2. Đối với cơ sở đóng gói theo hợp đồng, thư chỉ định và cho phép được đóng gói sơ cấp thuốc thành phẩm tại địa điểm mới có nêu cụ thể về công đoạn đóng gói được tiến hành tại địa điểm mới của cơ sở đóng gói. 3. Đối với thuốc vô khuẩn, kế hoạch thẩm định và/hoặc báo cáo thẩm định quy trình đóng gói sơ cấp thuốc thành phẩm tại địa điểm mới. 4. Số liệu độ ổn định của thuốc thành phẩm sau khi có thay đổi và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 5. Nghiên cứu ảnh hưởng của thời gian lưu trữ đối với bán thành phẩm ở dạng bulk product trong quá trình bảo quản và vận chuyển từ nơi sản xuất bán thành phẩm đến nơi đóng gói sơ cấp mới. |
|
|
|
|
|
|
| Nội dung thay đổi 6 (MaV-6) | Thay đổi tiêu chuẩn chất lượng dược chất [khi không có giấy chứng nhận tuân thủ dược điển châu Âu (CEP)] và/ hoặc thuốc thành phẩm thuộc các trường hợp sau: a) Mở rộng giới hạn của chỉ tiêu chất lượng; b) Bỏ bớt chỉ tiêu chất lượng và mức chất lượng. | x | x | x (không áp dụng với mục (b) vì không có tài liệu liên quan bào chế) |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Quy trình phân tích không thay đổi hoặc thay đổi rất ít (không cần thiết phải tiến hành thẩm định lại quy trình phân tích). 2. Không áp dụng đối với các trường hợp tiêu chuẩn chất lượng đã được duyệt của dược chất/ thuốc thành phẩm là tiêu chuẩn theo dược điển tham chiếu. 3. Tham khảo MiV-PA12 nếu thay đổi này dẫn đến việc xem xét lại CEP. 4. Thay đổi này không phải là kết quả của những tác động ngoài dự kiến xảy ra trong quá trình sản xuất hoặc liên quan đến độ ổn định. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | (a) Mở rộng giới hạn của chỉ tiêu chất lượng 1. Giải trình về lý do đề nghị thay đổi kèm theo các dữ liệu khoa học để chứng minh. 2. Bảng so sánh sự thay đổi giữa tiêu chuẩn chất lượng mới của dược chất/thuốc thành phẩm và tiêu chuẩn chất lượng đã được duyệt của dược chất/ thuốc thành phẩm có in đậm các nội dung thay đổi. 3. Tiêu chuẩn chất lượng mới của dược chất/thuốc thành phẩm. 4. Số liệu phân tích lô của dược chất /thuốc thành phẩm đối với tất cả các chỉ tiêu chất lượng trong tiêu chuẩn chất lượng mới tiến hành trên 02 lô (lô pilot hoặc lô sản xuất). 5. Số liệu độ ổn định của dược chất/ thuốc thành phẩm theo tiêu chuẩn chất lượng mới (đối với dược chất) và tiêu chuẩn chất lượng lưu hành mới (đối với thuốc thành phẩm) và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng đồng thời đề xuất biện pháp xử lý phù hợp. (b) Bỏ bớt chỉ tiêu chất lượng và mức chất lượng 1. Cung cấp các tài liệu từ D1- D4. 2. Phiếu kiểm nghiệm dược chất/thuốc thành phẩm theo tiêu chuẩn chất lượng mới (đối với dược chất) và tiêu chuẩn chất lượng lưu hành mới (đối với thuốc thành phẩm). |
|
|
|
|
|
|
| Nội dung thay đổi 7 (MaV-7) | Bổ sung, thay đổi cỡ lô đối với thuốc thành phẩm vô khuẩn | x |
| x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Thay đổi này không ảnh hưởng đến tính ổn định của quy trình sản xuất. 2. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi. 3. Công thức bào chế thuốc thành phẩm không thay đổi. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Kế hoạch thẩm định quy trình sản xuất và/hoặc báo cáo thẩm định quy trình sản xuất của ít nhất 03 lô sản xuất theo cỡ lô mới. 2. Bảng so sánh công thức lô sản xuất giữa cỡ lô đã được duyệt và cỡ lô mới. 3. Số liệu phân tích lô (dạng bảng so sánh) của ít nhất 02 lô sản xuất của thuốc thành phẩm trước và sau khi có thay đổi. 4. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. 5. Số liệu độ ổn định của thuốc thành phẩm sau khi có thay đổi và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành, đồng thời đề xuất biện pháp xử lý phù hợp. |
|
|
|
|
|
|
| Nội dung thay đổi 8 (MaV-8) | Bổ sung, thay đổi cỡ lô đối với thuốc thành phẩm không vô khuẩn | x |
| x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Áp dụng đối với thay đổi cỡ lô lớn hơn 10 lần so với cỡ lô đã được duyệt. Đối với thay đổi cỡ lô dưới hoặc bằng 10 lần cỡ lô đã được duyệt, xem MiV-PA13. 2. Thay đổi này không ảnh hưởng đến tính ổn định của quy trình sản xuất. 3. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Kế hoạch thẩm định quy trình và/hoặc báo cáo thẩm định quy trình sản xuất của ít nhất 03 lô sản xuất theo cỡ lô mới. 2. Bảng so sánh công thức lô sản xuất giữa cỡ lô đã được duyệt và cỡ lô mới. 3. Số liệu phân tích lô (dạng bảng so sánh) giữa ít nhất 01 lô sản xuất thuốc thành phẩm sau khi có thay đổi và 01 lô sản xuất thuốc thành phẩm trước khi có thay đổi kèm theo thư cam kết của cơ sở sản xuất thuốc thành phẩm và cơ sở đăng ký sẽ nộp dữ liệu phân tích lô cho một lô sản xuất đầy đủ kế tiếp. 4. Số liệu độ ổn định của thuốc thành phẩm sau khi có thay đổi và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 5. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. 6. Đối với các dạng bào chế rắn dùng đường uống: Số liệu so sánh biểu đồ hòa tan trên ít nhất 01 lô sản xuất giữa các thuốc thành phẩm trước và sau khi có thay đổi. |
|
|
|
|
|
|
| Nội dung thay đổi 9 (MaV-9) | Thay đổi lớn trong quy trình sản xuất thuốc thành phẩm |
|
|
|
|
|
|
| TH1 | Các thuốc có chứng minh TĐSH | x | x | x |
|
| x |
| TH2 | Các thuốc không có chứng minh TĐSH | x | x | x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Địa điểm sản xuất không thay đổi. Nếu thay đổi địa điểm sản xuất, áp dụng thêm MaV-4. 2. Thay đổi này không ảnh hưởng tiêu cực đến chất lượng, an toàn và hiệu quả của thuốc. 3. Thay đổi nhỏ trong quy trình sản xuất thuốc thành phẩm không vô khuẩn áp dụng theo MiV-PA20. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Mô tả quy trình sản xuất mới và giải trình về mặt kỹ thuật các lý do đề nghị thay đổi. 2. Kế hoạch thẩm định quy trình và/hoặc báo cáo thẩm định quy trình sản xuất mới. 3. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. Hoặc, có thể thay bằng tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành dự kiến của thuốc thành phẩm để chứng minh thuốc được sản xuất theo quy trình sản xuất mới ít nhất là tương đương hoặc tốt hơn về mặt chất lượng, an toàn và hiệu quả so với thuốc sản xuất theo quy trình sản xuất cũ. 4. Bảng so sánh số liệu phân tích lô trên tối thiểu 01 lô sản xuất giữa các thuốc thành phẩm trước và sau khi có thay đổi. 5. Số liệu độ ổn định của thuốc thành phẩm sau khi có thay đổi và báo cáo nếu có bất cứ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 6. Đối với các dạng bào chế rắn dùng đường uống: Số liệu so sánh biểu đồ hòa tan giữa các thuốc thành phẩm trước và sau khi có thay đổi. 7. Giải trình các căn cứ khoa học của việc không nộp số liệu nghiên cứu tương đương sinh học của thuốc thành phẩm sau khi có thay đổi (đối với các thuốc đã được cấp số đăng ký là thuốc có chứng minh tương đương sinh học). |
|
|
|
|
|
|
| Nội dung thay đổi 10 (MaV-10) | Thay đổi loại hoặc lượng tá dược a) Đối với dạng bào chế giải phóng ngay dùng đường uống (ở mức 2 và 3, Phần III (Thành phần và công thức)- hướng dẫn SUPAC- IR); b) Đối với dạng bào chế giải phóng biến đổi dùng đường uống; c) Đối với các dạng bào chế đặc biệt khác như các chế phẩm vô khuẩn. |
|
|
|
|
|
|
| TH1 | Các thuốc có chứng minh TĐSH | x | x | x |
|
| x |
| TH2 | Các thuốc không có chứng minh TĐSH | x | x | x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Các chỉ tiêu chất lượng và mức chất lượng tương ứng trong tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi, trừ việc cập nhật thông tin về mô tả hình thức mới của thuốc (nếu có thay đổi hình thức thuốc). 2. Số liệu so sánh biểu đồ hòa tan giữa các thuốc thành phẩm trước và sau khi thay đổi đạt quy định theo SUPAC-IR hoặc SUPAC-MR. 3. Thay thế một tá dược bằng một tá dược khác tương đương về các đặc tính sử dụng (có cùng chức năng). 4. Đối với những thay đổi khác về loại hoặc lượng tá dược của dạng bào chế giải phóng ngay dùng đường uống và các dạng bào chế không đặc biệt khác, xem MiV-PA15. Trường hợp thay đổi loại tá dược, nhà sản xuất tá dược phải có thêm: 5. Cơ sở sản xuất tá dược đáp ứng thực hành tốt sản xuất nguyên liệu làm thuốc (GMP) (thực hiện theo lộ trình quy định). |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Giải trình về đề nghị thay đổi bằng tài liệu phát triển dược học phù hợp. 2. Số liệu độ ổn định của thuốc thành phẩm sau khi thay đổi và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành, đồng thời đề xuất biện pháp xử lý phù hợp. 3. Đối với các thuốc có dạng bào chế rắn dùng đường uống: Số liệu so sánh biểu đồ hòa tan trên ít nhất 01 lô pilot/ lô sản xuất đại diện giữa các thuốc thành phẩm trước và sau khi có thay đổi. 4. Giải trình các căn cứ khoa học của việc không nộp số liệu nghiên cứu tương đương sinh học của thuốc thành phẩm sau khi có thay đổi (đối với các thuốc đã được cấp số đăng ký là thuốc có chứng minh tương đương sinh học) 5. Bảng so sánh công thức bào chế đã được duyệt lần đầu tiên và công thức bào chế đề nghị thay đổi có in đậm các nội dung thay đổi, trong đó lượng từng thành phần được thể hiện dưới dạng phần trăm khối lượng trên tổng khối lượng các thành phần trong công thức 6. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành của thuốc thành phẩm sau khi có thay đổi trong đó đã cập nhật các thay đổi về mô tả hình thức mới của thuốc (trong trường hợp có thay đổi mô tả hình thức thuốc). 7. Số liệu phân tích lô (dạng bảng so sánh) trên ít nhất 02 lô sản xuất (hoặc 01 lô sản xuất và 02 lô pilot) giữa các thuốc thành phẩm trước và sau khi thay đổi. 8. Tiêu chuẩn chất lượng của tá dược mới (nếu khi thay đổi có sử dụng tá dược chưa có trong công thức bào chế đã được duyệt). 9. Đối với tá dược có nguồn gốc từ động vật nhai lại, yêu cầu nộp giấy chứng nhận không có TSE hoặc BSE do cơ quan quản lý thú ý có thẩm quyền cấp. 10. Công thức lô sản xuất mới. 11. Kế hoạch thẩm định và/hoặc báo cáo thẩm định quy trình sản xuất thuốc thành phẩm sau khi thay đổi trên ít nhất 03 lô sản xuất. 12. Các phần từ P3.1- P3.4 theo ACTD đã cập nhật các thay đổi phù hợp với công thức bào chế mới. 13. Giải trình phù hợp kèm theo các số liệu thực nghiệm chứng minh (nếu cần) về việc các thay đổi trong công thức bào chế này không làm ảnh hưởng đến kết quả của các quy trình phân tích trong tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. Nếu thay đổi trong công thức bào chế này dẫn đến việc phải thay đổi quy trình phân tích trong tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm, yêu cầu tham chiếu MiV-PA27 để bổ sung các tài liệu theo quy định. 14. Đối với những thay đổi về loại và lượng chất bảo quản, kết quả của chỉ tiêu hiệu quả bảo quản (PET) ở nồng độ chất bảo quản thấp nhất (nếu có). 15. Giấy tờ pháp lý của cơ sở sản xuất tá dược mới theo quy định tại khoản 11 Điều 22 Thông tư này. Không yêu cầu tài liệu này đối với tá dược đã có Giấy đăng ký lưu hành tại Việt Nam. |
|
|
|
|
|
|
| Nội dung thay đổi 11 (MaV-11) | Thay đổi khối lượng màng bao viên nén hoặc khối lượng và/hoặc kích cỡ vỏ nang đối với thuốc giải phóng biến đổi dùng đường uống |
|
|
|
|
|
|
| TH1 | Các thuốc có chứng minh TĐSH | x | x | x |
|
| x |
| TH2 | Các thuốc không có chứng minh TĐSH | x | x | x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Số liệu so sánh biểu đồ hòa tan giữa thuốc thành phẩm trước và sau khi thay đổi đạt yêu cầu theo SUPAC-MR. 2. Các chỉ tiêu chất lượng và mức chất lượng tương ứng trong tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi, trừ việc cập nhật thông tin về mô tả hình thức mới của thuốc (nếu có thay đổi hình thức thuốc) 3. Thay đổi khối lượng màng bao viên nén hoặc khối lượng và/hoặc kích cỡ vỏ nang đối với dạng thuốc rắn giải phóng ngay dùng đường uống, xem MiV-PA16. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Số liệu so sánh biểu đồ hòa tan trên ít nhất 01 lô pilot/lô sản xuất giữa các thuốc thành phẩm trước và sau khi có thay đổi. 2. Giải trình các căn cứ khoa học của việc không nộp số liệu nghiên cứu tương đương sinh học của thuốc thành phẩm sau khi có thay đổi (đối với các thuốc đã được cấp số đăng ký là thuốc có chứng minh tương đương sinh học). 3. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành của thuốc thành phẩm sau khi thay đổi trong đó đã cập nhật các thay đổi về mô tả hình thức mới của thuốc (trong trường hợp có thay đổi mô tả hình thức thuốc). 4. Cam kết của cơ sở sản xuất thuốc thành phẩm và cơ sở đăng ký về việc thay đổi này không làm ảnh hưởng đến quy trình phân tích trong tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. 5. Công thức bào chế cho 01 đơn vị liều và công thức lô sản xuất đã được duyệt và sau khi có thay đổi. 6. Số liệu độ ổn định của thuốc thành phẩm sau khi có thay đổi và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 7. Phần P3.1- P3.4 theo ACTD đã cập nhật các thay đổi phù hợp với công thức bào chế mới. |
|
|
|
|
|
|
| Nội dung thay đổi 12 (MaV- 12) | Thay đổi liên quan đến bao bì đóng gói sơ cấp của thuốc thành phẩm vô khuẩn áp dụng đối với các trường hợp sau: a) Thay đổi thành phần và lượng các thành phần trong chất liệu bao bì đóng gói đã được duyệt và/ hoặc b) Thay đổi loại bao bì đóng gói và/ hoặc c) Bổ sung thêm chất liệu bao bì đóng gói. | x |
| x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi. 2. Thay đổi liên quan đến bao bì đóng gói sơ cấp đối với thuốc thành phẩm không vô khuẩn, xem MiV-PA28. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Kế hoạch thẩm định và/hoặc báo cáo thẩm định quy trình sản xuất và quy trình tiệt khuẩn khi sử dụng bao bì đóng gói sơ cấp mới trong sản xuất thuốc thành phẩm. 2. Số liệu độ ổn định của thuốc thành phẩm trong bao bì đóng gói sơ cấp mới và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 3. Bằng chứng chứng minh không có sự tương tác giữa thuốc và bao bì đóng gói sơ cấp mới. 4. Bảng so sánh về tiêu chuẩn chất lượng giữa bao bì đóng gói sơ cấp mới và bao bì đóng gói sơ cấp đã được duyệt. 5. Các số liệu khoa học chứng minh sự phù hợp của việc sử dụng bao bì đóng gói sơ cấp mới (số liệu so sánh về tính thấm giữa bao bì đóng gói sơ cấp mới và bao bì đóng gói sơ cấp đã được duyệt đối với các thông số hàm ẩm, O2, CO2) 6. Các phần P3 và P7 theo ACTD đã cập nhật các nội dung thay đổi tương ứng. 7. Tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm 8. Mẫu nhãn đã được duyệt và mẫu nhãn mới (nếu có thay đổi). |
|
|
|
|
|
|
| Nội dung thay đổi 13 (MaV-13) | Thay đổi hoặc bổ sung quy cách đóng gói thuốc thành phẩm trong bao bì sơ cấp và, hoặc thay đổi hình dạng hoặc kích thước đồ bao gói sơ cấp hoặc hệ thống đóng kín đối với thuốc vô khuẩn dạng rắn và dạng dung dịch. | x |
| x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Không ảnh hưởng đến tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm, ngoại trừ thay đổi trong mô tả quy cách đóng gói của thuốc thành phẩm. 2. Quy cách đóng gói mới phù hợp với liều dùng và thời gian sử dụng đã được duyệt trong hướng dẫn sử dụng. 3. Chất liệu bao bì đóng gói không thay đổi. 4. Thay đổi hoặc bổ sung quy cách đóng gói thuốc thành phẩm trong bao bì sơ cấp và/hoặc thay đổi hình dạng hoặc kích thước đồ bao gói sơ cấp hoặc hệ thống đóng kín của thuốc không vô khuẩn, xem MiV-PA30. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Chứng minh sự phù hợp giữa quy cách đóng gói mới với liều dùng và thời gian sử dụng đã được duyệt trong hướng dẫn sử dụng. 2. Số liệu thẩm định quy trình sản xuất, hệ thống đóng kín và tiệt khuẩn. 3. Số liệu độ ổn định của thuốc thành phẩm trong các quy cách đóng gói sơ cấp mới và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 4. Tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm 5. Mẫu nhãn đã được duyệt và mẫu nhãn mới (nếu có thay đổi/ bổ sung). |
|
|
|
|
|
|
| Nội dung thay đổi 14 (MaV-14) | Bổ sung hoặc thay thế dung môi, dung dịch hòa tan/ phân tán/ pha loãng thuốc | x | x | x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Thay đổi này không gây ra bất kỳ thay đổi nào về dạng bào chế, liều dùng, chỉ định, cách dùng của thuốc. 2. Bỏ bớt dung môi, dung dịch hòa tan/ phân tán/pha loãng thuốc, xem MiV-PA18. 3. Nếu có thay đổi hạn dùng và/hoặc điều kiện bảo quản thuốc sau khi mở nắp và/hoặc sau khi pha với các dung môi, dung dịch mới, xem MaV-15/MiV-PA34 và/hoặc MaV-16/MiV- PA35 để cung cấp các tài liệu phù hợp với từng loại thay đổi tương ứng. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Nộp toàn bộ phần P theo ACTD đối với dung dịch hòa tan/ phân tán/ pha loãng mới và nộp phần S theo ACTD đối với các thành phần có tác dụng dược lý trong dung dịch hòa tan/ phân tán/ pha loãng mới nếu đây là các thành phần chưa có trong công thức bào chế của các dung dịch hòa tan/phân tán/ pha loãng đã được duyệt. Nộp tiêu chuẩn chất lượng của dung môi mới (nếu có sử dụng dung môi mới). 2. Tài liệu pháp lý chứng minh cơ sở sản xuất/địa điểm sản xuất dung môi/ dung dịch hòa tan/ phân tán/ pha loãng mới đạt GMP phù hợp với các dạng bào chế này (nếu các dung môi, dung dịch hòa tan/ phân tán/ pha loãng mới được sản xuất tại cơ sở sản xuất/ địa điểm sản xuất mới). 3. Mẫu nhãn mới và mẫu nhãn đã được duyệt (nếu có thay đổi/ bổ sung). |
|
|
|
|
|
|
| Nội dung thay đổi 15 (MaV-15) | Tăng hạn dùng thuốc thành phẩm: a) Khi đóng gói trong bao bì sơ cấp theo các quy cách đóng gói đã được duyệt và/ hoặc b) Sau khi mở nắp lần đầu và/ hoặc c) Sau khi hòa tan, phân tán hoặc pha loãng. | x |
| x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Đối với (a) & (b) - Các nghiên cứu phải thể hiện được sự phù hợp với tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. 2. Đối với (c) - Các nghiên cứu phải thể hiện được sự phù hợp với tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm sau khi hòa tan, phân tán hoặc pha loãng. 3. Đối với trường hợp giảm hạn dùng thuốc thành phẩm, xem MiV-PA34. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Số liệu độ ổn định dài hạn của thuốc thành phẩm phù hợp với hạn dùng mới tương ứng cho ít nhất 02 lô (lô pilot hoặc lô sản xuất) kèm theo các kết quả thử nghiệm vi sinh vật phù hợp trong các trường hợp: a) Khi đóng gói trong bao bì sơ cấp theo các quy cách đóng gói đã được duyệt và/hoặc b) Sau khi mở nắp lần đầu và/hoặc c) Sau khi hòa tan, phân tán hoặc pha loãng 2. Thuyết minh về mặt kỹ thuật các lý do cho đề nghị thay đổi này. 3. Tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm |
|
|
|
|
|
|
| Nội dung thay đổi 16 (MaV-16) | Thay đổi điều kiện bảo quản thuốc thành phẩm (ít khắc nghiệt hơn điều kiện bảo quản đã phê duyệt): a) Khi đóng gói trong bao bì sơ cấp theo các quy cách đóng gói đã được duyệt và/hoặc b) Sau khi mở nắp lần đầu và/hoặc c) Sau khi hòa tan, phân tán hoặc pha loãng. | x |
| x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Đối với (a) & (b) - Các nghiên cứu phải thể hiện được sự phù hợp với tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. 2. Đối với (c)- Các nghiên cứu phải thể hiện được sự phù hợp với tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm sau khi hòa tan, phân tán hoặc pha loãng. 3. Đối với thay đổi điều kiện bảo quản thuốc thành phẩm (khắc nghiệt hơn so với điều kiện bảo quản đã được chấp thuận), xem MiV- PA35. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Số liệu độ ổn định dài hạn của thuốc thành phẩm ở điều kiện bảo quản mới phù hợp với hạn dùng đã được duyệt tương ứng của ít nhất 02 lô (lô pilot hoặc lô sản xuất) kèm theo các kết quả thử nghiệm vi sinh vật phù hợp trong các trường hợp: a) Khi đóng gói trong bao bì sơ cấp theo các quy cách đóng gói đã được duyệt và/hoặc b) Sau khi mở nắp lần đầu và/hoặc c) Sau khi hòa tan, phân tán hoặc pha loãng 2. Thuyết minh về mặt kỹ thuật các lý do cho đề nghị thay đổi này. 3. Tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm |
|
|
|
|
|
|
| Nội dung thay đổi 17 (MaV-17) | Thay đổi lớn trong quy trình sản xuất dược chất (khi không có Giấy chứng nhận tuân thủ Dược điển châu Âu (CEP) | x | x | x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Không dẫn đến các thay đổi bất lợi về loại và/hoặc lượng tạp chất đòi hỏi phải có những nghiên cứu thêm về tính an toàn 2. Phương pháp tổng hợp có sự thay đổi. Trường hợp phương pháp tổng hợp dược chất không thay đổi, áp dụng MiV-PA7. 3. Quy trình sản xuất dược chất không sử dụng bất cứ nguyên liệu nào có nguồn gốc từ người/động vật đòi hỏi phải có đánh giá độ an toàn về nhiễm vi rút hoặc có giải trình phù hợp. 4. Chỉ tiêu lý hóa và các chỉ tiêu liên quan khác của dược chất không thay đổi. 5. Độ ổn định của dược chất không thay đổi. 6. Nếu có thay đổi liên quan tiêu chuẩn chất lượng dược chất, áp dụng thêm MiV-PA8. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Hồ sơ tổng thể của dược chất (DMF) hoặc phần hồ sơ cập nhật của dược chất theo ACTD hoặc tài liệu kiểm tra tương đương được cung cấp từ một trong các nước tham chiếu quy định tại Thông tư đăng ký thuốc. 2. Bảng so sánh quy trình sản xuất đã được duyệt và quy trình sản xuất mới có in đậm những nội dung thay đổi. 3. Đối với dược chất vô khuẩn, nộp báo cáo thẩm định quy trình sản xuất mới của dược chất. 4. Thư của cơ sở sản xuất dược chất tuyên bố không xuất hiện tạp chất mới bằng hoặc vượt ngưỡng chấp nhận cũng như không có sự tăng giới hạn tạp chất đòi hỏi phải có nghiên cứu thêm về tính an toàn. 5. Thư của cơ sở sản xuất dược chất tuyên bố không có sự thay đổi trong tiêu chuẩn chất lượng đã được duyệt của dược chất hoặc nếu có bất cứ thay đổi nào trong tiêu chuẩn chất lượng dược chất (ví dụ theo hướng chặt chẽ hơn) thì cần nộp bảng so sánh giữa tiêu chuẩn chất lượng mới và tiêu chuẩn chất lượng đã được duyệt của dược chất. 6. Phiếu kiểm nghiệm và/hoặc bảng so sánh dữ liệu phân tích lô đối với ít nhất hai lô (lô pilot hoặc lô sản xuất) giữa thuốc thành phẩm sử dụng dược chất sản xuất theo quy trình sản xuất mới và thuốc thành phẩm sử dụng dược chất sản xuất theo quy trình sản xuất đã được duyệt. 7. Cam kết của cơ sở sản xuất thuốc thành phẩm về việc đã bắt đầu tiến hành và sẽ hoàn thành nghiên cứu độ ổn định của thuốc thành phẩm được sản xuất từ dược chất sản xuất theo quy trình sản xuất mới, và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 8. Phiếu kiểm nghiệm và/hoặc bảng so sánh dữ liệu phân tích lô đối với ít nhất hai lô dược chất được sản xuất theo quy trình sản xuất mới. |
|
|
|
|
|
|
II. THAY ĐỔI NHỎ CẦN PHÊ DUYỆT TRƯỚC KHI THỰC HIỆN
| Nội dung thay đổi | Pháp chế | Tiêu chuẩn | Bào chế | Dược lý | Lâm sàng | BA/BE | |
| Nội dung thay đổi 1 (MiV- PA1) | Thay đổi tên thuốc thành phẩm | x |
|
|
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Chỉ thay đổi tên thuốc thành phẩm, các phần khác không thay đổi (công thức, tiêu chuẩn chất lượng, nguồn nguyên liệu, quy trình sản xuất…). 2. Tên mới tuân thủ theo quy định về đặt tên thuốc tại Thông tư quy định việc đăng ký thuốc và Thông tư quy định về ghi nhãn thuốc. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Giấy chứng nhận dược phẩm (CPP) có tên thuốc mới đối với thuốc sản xuất tại nước ngoài. |
|
|
|
|
|
|
| Nội dung thay đổi 2 (MiV- PA2) | Thay đổi/bổ sung nội dung của hướng dẫn sử dụng và/hoặc mẫu nhãn áp dụng đối với các trường hợp sau: a) Thay đổi thiết kế mà không thay đổi ý nghĩa; b) Bổ sung/bỏ bớt/thay thế hình ảnh, sơ đồ, hoặc nội dung; c) Bổ sung/thay đổi theo hướng chặt chẽ hơn cảnh báo, thận trọng, chống chỉ định và/hoặc tác dụng không mong muốn so với hướng dẫn sử dụng đã được duyệt; d) Thu hẹp nhóm bệnh nhân sử dụng; Bỏ bớt chỉ định. |
|
|
|
|
|
|
| TH1 | Có thay đổi nội dung hướng dẫn sử dụng kèm hồ sơ lâm sàng chứng minh | x |
|
| x | x |
|
| TH2 | Có thay đổi nội dung hướng dẫn sử dụng không kèm hồ sơ lâm sàng | x |
|
| x |
|
|
| TH3 | Không thay đổi nội dung hướng dẫn sử dụng thuốc | x |
|
|
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Thay đổi không phải là thay đổi lớn và không chứa nội dung quảng cáo, không thuộc quy định tại khoản 2 Điều 35 Thông tư số 01/2018/TT- BYT ngày 18/01/2018 của Bộ Y tế hướng dẫn ghi nhãn thuốc. 2. Đối với thay đổi lớn về mẫu nhãn/hướng dẫn sử dụng, xem MaV-1 và MaV-2. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Hướng dẫn sử dụng và/hoặc mẫu nhãn mới. Bảng so sánh chi tiết các nội dung thay đổi của tờ hướng dẫn sử dụng (trường hợp thay đổi nội dung tờ hướng dẫn sử dụng). 2. Giải trình lý do thay đổi. 3. Tài liệu tham khảo cho nội dung thay đổi (nếu có). |
|
|
|
|
|
|
| Nội dung thay đổi 3 (MiV- PA3) | Thay đổi cơ sở/địa điểm cơ sở chịu trách nhiệm xuất xưởng lô | x | x |
|
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Chỉ áp dụng cho cơ sở xuất xưởng lô. 2. Đã hoàn thành việc chuyển giao quy trình phân tích từ phòng kiểm nghiệm/ cơ sở xuất xưởng lô cũ sang phòng kiểm nghiệm/ cơ sở xuất xưởng lô mới. 3. Cơ sở sản xuất thuốc thành phẩm không thay đổi. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Tài liệu pháp lý chứng minh cơ sở/địa điểm xuất xưởng lô mới được cơ quan có thẩm quyền cho phép xuất xưởng lô như: giấy chứng nhận GMP hoặc GLP, hoặc CPP có chứng nhận GMP hoặc GLP cho cơ sở/địa điểm xuất xưởng lô mới. 2. Số liệu thẩm tra quy trình phân tích đã được duyệt tại địa điểm xuất xưởng lô mới hoặc hồ sơ chuyển giao quy trình phân tích đã được duyệt từ địa điểm xuất xưởng lô đã được duyệt sang địa điểm xuất xưởng lô mới (nếu có). 3. Giấy ủy quyền từ chủ sở hữu sản phẩm ủy quyền cơ sở xuất xưởng lô mới thực hiện xuất xưởng lô (nếu có). |
|
|
|
|
|
|
| Nội dung thay đổi 4 (MiV- PA4) | Thay đổi và/hoặc bổ sung cơ sở sản xuất/địa điểm sản xuất của dược chất [khi có giấy chứng nhận tuân thủ dược điển châu Âu (CEP)] |
|
|
|
|
|
|
| TH1 | Các thuốc có chứng minh TĐSH | x | x | x |
|
| x |
| TH2 | Các thuốc không có chứng minh TĐSH | x | x | x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Tiêu chuẩn chất lượng dược chất không thay đổi. 2. Thay đổi và/hoặc bổ sung cơ sở sản xuất/ địa điểm sản xuất dược chất khi không có CEP, xem MaV-3. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Giấy chứng nhận tuân thủ dược điển châu Âu (CEP) đối với dược chất bản mới nhất còn hiệu lực kèm theo tất cả các phụ lục được ban hành bởi Hội đồng châu Âu về chất lượng thuốc (EDQM). 2. Số liệu phân tích lô (dạng bảng so sánh) của ít nhất 02 lô pilot giữa dược chất sản xuất tại địa điểm mới và dược chất sản xuất tại địa điểm đã được duyệt. Đối với trường hợp thuốc được công bố có chứng minh tương đương sinh học, bảng so sánh phân tích lô phải bao gồm cả các chỉ tiêu chất lượng có trong tiêu chuẩn chất lượng đã được duyệt của dược chất và các chỉ tiêu chất lượng không có trong tiêu chuẩn nhưng đã được thiết lập trong quá trình nghiên cứu phát triển thành phẩm (là các chỉ tiêu đã được mô tả tại phần P2. Phát triển dược học trong hồ sơ đăng ký thuốc, ví dụ: kích thước tiểu phân, dạng thù hình, trạng thái hydrat hoá/solvat hoá, đặc tính hòa tan, khối lượng riêng biểu kiến, độ trơn chảy của dược chất) kèm theo tài liệu chứng minh nếu cần. 3. Số liệu so sánh biểu đồ hòa tan giữa các thuốc thành phẩm trước và sau khi thay đổi đối với trường hợp thuốc đã được công bố có tài liệu chứng minh tương đương sinh học. 4. Nếu thời hạn phải kiểm tra lại chất lượng dược chất không nêu trong CEP, nộp số liệu nghiên cứu độ ổn định của dược chất sản xuất tại địa điểm mới ở điều kiện dài hạn và điều kiện lão hóa cấp tốc trên 02 lô pilot phù hợp với thời hạn phải kiểm tra lại chất lượng dược chất theo đề xuất của cơ sở sản xuất dược chất. 5. Cam kết của cơ sở sản xuất thuốc thành phẩm về việc tiến hành nghiên cứu độ ổn định của thuốc thành phẩm được sản xuất từ dược chất sản xuất tại địa điểm mới ở điều kiện dài hạn và điều kiện lão hóa cấp tốc, và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. |
|
|
|
|
|
|
| Nội dung thay đổi 5 (MiV- PA5) | Thay đổi cỡ lô sản xuất dược chất [khi không có giấy chứng nhận tuân thủ dược điển châu Âu (CEP)] | x |
| x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Thay đổi này không ảnh hưởng đến tính ổn định của quy trình sản xuất. 2. Tiêu chuẩn chất lượng dược chất không thay đổi. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Số liệu phân tích lô (dạng bảng so sánh) theo tiêu chuẩn chất lượng dược chất đã được duyệt của ít nhất 01 lô sản xuất hoặc 01 lô pilot giữa dược chất sản xuất theo cỡ lô mới và dược chất sản xuất theo cỡ lô đã được duyệt. Số liệu phân tích lô cho 02 lô sản xuất đầy đủ kế tiếp phải sẵn có để nộp khi có yêu cầu. Trong quá trình phân tích lô, nếu có kết quả nào không đạt tiêu chuẩn chất lượng phải có báo cáo đồng thời đề xuất biện pháp xử lý phù hợp. 2. Cam kết của cơ sở sản xuất dược chất về việc tiêu chuẩn chất lượng của dược chất không thay đổi và tính ổn định của quy trình sản xuất dược chất không bị ảnh hưởng. 3. Cập nhật mục S theo ACTD. |
|
|
|
|
|
|
| Nội dung thay đổi 6 (MiV- PA6) | Thay đổi trong kiểm soát trong quá trình sản xuất đối với dược chất (bao gồm theo hướng chặt chẽ hơn, bổ sung chỉ tiêu kiểm soát mới và khi không có giấy chứng nhận tuân thủ dược điển châu Âu CEP) | x | x | x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Giới hạn chỉ tiêu kiểm soát trong quy trình sản xuất chặt chẽ hơn hoặc bổ sung chỉ tiêu kiểm soát mới. 2. Thay đổi này không phải là hệ quả của bất kỳ cam kết nào về việc xem xét lại giới hạn trong tiêu chuẩn chất lượng từ lần thẩm định trước. 3. Thay đổi này không phải là kết quả của biến cố ngoại ý phát sinh trong quá trình sản xuất như: tạp chất mới chưa xác định, thay đổi giới hạn tổng tạp chất... Quy trình phân tích của chỉ tiêu kiểm soát mới không liên quan đến một kỹ thuật mới không theo chuẩn hoặc một kỹ thuật cơ bản được sử dụng theo cách mới. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Mô tả quy trình phân tích và hồ sơ thẩm định quy trình phân tích của tất cả các quy trình phân tích mới. 2. Bảng so sánh sự thay đổi về kiểm soát trong quy trình sản xuất có in đậm những nội dung thay đổi liên quan. 3. So sánh số liệu phân tích lô trên 02 lô sản xuất của dược chất đối với tất cả các chỉ tiêu chất lượng trong tiêu chuẩn chất lượng . |
|
|
|
|
|
|
| Nội dung thay đổi 7 (MiV- PA7) | Thay đổi trong quy trình sản xuất dược chất [khi không có giấy chứng nhận tuân thủ dược điển châu Âu (CEP)] | x | x | x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Không dẫn đến các thay đổi bất lợi về loại và/hoặc lượng tạp chất đòi hỏi phải có những nghiên cứu thêm về tính an toàn. 2. Độ ổn định của dược chất không thay đổi. 3. Phương pháp tổng hợp dược chất không thay đổi (ví dụ: chất trung gian không thay đổi). 4. Quy trình sản xuất dược chất không sử dụng bất cứ nguyên liệu nào có nguồn gốc từ người/động vật đòi hỏi phải có đánh giá độ an toàn về nhiễm vi rút. Chỉ tiêu lý hóa và các chỉ tiêu liên quan khác của dược chất không thay đổi. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Hồ sơ tổng thể của dược chất (DMF) hoặc phần hồ sơ cập nhật của dược chất theo ACTD hoặc tài liệu kiểm tra tương đương được cung cấp từ một trong các nước tham chiếu quy định tại Thông tư đăng ký thuốc. 2. Bảng so sánh quy trình sản xuất đã được duyệt và quy trình sản xuất mới có in đậm những nội dung thay đổi. 3. Phiếu kiểm nghiệm của 02 lô dược chất được sản xuất theo quy trình sản xuất mới. 4. Bảng so sánh số liệu phân tích lô của ít nhất 02 lô (lô pilot hoặc lô sản xuất) giữa thuốc thành phẩm sử dụng dược chất sản xuất theo quy trình sản xuất mới và thuốc thành phẩm sử dụng dược chất sản xuất theo quy trình sản xuất đã được duyệt. 5. Thư của cơ sở sản xuất dược chất tuyên bố không xuất hiện tạp chất mới bằng hoặc vượt ngưỡng chấp nhận cũng như không có sự tăng giới hạn tạp chất đòi hỏi phải có nghiên cứu thêm về tính an toàn. 6. Thư của cơ sở sản xuất dược chất tuyên bố không có sự thay đổi trong tiêu chuẩn chất lượng đã được duyệt của dược chất hoặc nếu có bất cứ thay đổi nào trong tiêu chuẩn chất lượng dược chất (ví dụ theo hướng chặt chẽ hơn) thì cần nộp bảng so sánh giữa tiêu chuẩn chất lượng mới và tiêu chuẩn chất lượng đã được duyệt của dược chất. 7. Cam kết của cơ sở sản xuất thuốc thành phẩm về việc đã bắt đầu tiến hành và sẽ hoàn thành nghiên cứu độ ổn định của thuốc thành phẩm được sản xuất từ dược chất sản xuất theo quy trình sản xuất mới, và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 8. Đối với dược chất vô khuẩn, nộp báo cáo thẩm định quy trình sản xuất mới của dược chất. Nếu có thay đổi trong tiêu chuẩn chất lượng dược chất, yêu cầu cung cấp các mục S4-S5 theo ACTD đối với dược chất sản xuất theo quy trình sản xuất mới. |
|
|
|
|
|
|
| Nội dung thay đổi 8 (MiV-PA8) | Thay đổi tiêu chuẩn chất lượng dược chất áp dụng đối với các trường hợp sau: a) Mức giới hạn các chỉ tiêu chất lượng chặt chẽ hơn; b) Bổ sung thêm chỉ tiêu chất lượng và mức chất lượng mới. | x | x |
|
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Chỉ áp dụng cho những trường hợp tiêu chuẩn chất lượng đã được duyệt của dược chất không theo một trong các dược điển tham chiếu và những dược chất generic không có giấy chứng nhận tuân thủ dược điển châu Âu (CEP). 2. Đối với (b) - chỉ áp dụng cho các phương pháp không có trong dược điển. 3. Nếu thay đổi này dẫn đến thay đổi CEP, xem MiV-PA12. 4. Nếu mở rộng giới hạn các chỉ tiêu chất lượng và/ hoặc bỏ bớt chỉ tiêu chất lượng và mức chất lượng trong tiêu chuẩn chất lượng dược chất, xem MaV- 5. Thay đổi này không phải là kết quả của biến cố ngoại ý xuất hiện trong quá trình sản xuất hoặc do quan ngại về độ ổn định. 6. Quy trình phân tích của các chỉ tiêu chất lượng đã có trong tiêu chuẩn chất lượng đã được duyệt của dược chất không thay đổi hoặc thay đổi không đáng kể (không cần thiết phải tiến hành thẩm định lại quy trình phân tích). |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | (a) Mức giới hạn các chỉ tiêu chất lượng chặt chẽ hơn: 1. Bảng so sánh các chỉ tiêu chất lượng và các mức chất lượng tương ứng giữa tiêu chuẩn chất lượng trước và sau khi thay đổi của dược chất, có in đậm các nội dung thay đổi. 2. Bảng so sánh số liệu phân tích lô theo tiêu chuẩn chất lượng mới của dược chất trên 02 lô (lô pilot hoặc lô sản xuất). 3. Giải trình các lý do kỹ thuật dẫn đến đề nghị thay đổi này. (b) Bổ sung thêm chỉ tiêu chất lượng và mức chất lượng mới: Ngoài những tài liệu nêu trên cần bổ sung thêm: Mô tả quy trình phân tích mới và báo cáo số liệu thẩm định quy trình phân tích mới. |
|
|
|
|
|
|
| Nội dung thay đổi 9 (MiV- PA9) | Thay đổi quy trình phân tích trong tiêu chuẩn chất lượng dược chất của các dược chất chưa có trong dược điển | x | x |
|
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Kết quả thẩm định quy trình phân tích mới cho thấy quy trình phân tích mới ít nhất là tương đương với quy trình phân tích đã được duyệt. 2. Nếu thay đổi này dẫn đến thay đổi CEP, xem MiV-PA12. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Mô tả quy trình phân tích mới, báo cáo số liệu thẩm định quy trình phân tích mới và so sánh kết quả phân tích thu được từ quy trình phân tích mới và quy trình phân tích đã được duyệt. 2. Tiêu chuẩn chất lượng mới của dược chất đã cập nhật các thay đổi trong quy trình phân tích. 3. Phiếu kiểm nghiệm dược chất trên ít nhất 02 lô theo tiêu chuẩn chất lượng mới. |
|
|
|
|
|
|
| Nội dung thay đổi 10 (MiV- PA10) | Thay đổi hạn dùng hoặc thời hạn phải kiểm tra lại chất lượng dược chất | x |
| x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Các nghiên cứu độ ổn định phải thể hiện được sự phù hợp với tiêu chuẩn chất lượng đã được duyệt của dược chất 2. Không thay đổi điều kiện bảo quản. 3. Nếu thay đổi này dẫn đến thay đổi CEP, xem MiV-PA12. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Số liệu độ ổn định của dược chất trên ít nhất 02 lô (lô pilot hoặc lô sản xuất) phù hợp với hạn dùng mới hoặc thời hạn phải kiểm tra lại chất lượng mới của dược chất. 2. Tiêu chuẩn chất lượng đã được duyệt của dược chất. |
|
|
|
|
|
|
| Nội dung thay đổi 11 (MiV- PA11) | Thay đổi điều kiện bảo quản dược chất | x |
| x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Các nghiên cứu độ ổn định phải thể hiện được sự phù hợp với tiêu chuẩn chất lượng đã được duyệt của dược chất. 2. Không thay đổi hạn dùng/thời hạn phải kiểm tra lại chất lượng của dược chất. 3. Nếu thay đổi này dẫn đến thay đổi CEP, xem MiV-PA12. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Tiêu chuẩn chất lượng đã được duyệt của dược chất. 2. Số liệu độ ổn định của dược chất ở điều kiện bảo quản mới trên ít nhất 02 lô (lô pilot hoặc lô sản xuất) phù hợp với hạn dùng hoặc thời hạn phải kiểm tra lại chất lượng đã được duyệt của dược chất. |
|
|
|
|
|
|
| Nội dung thay đổi 12 (MiV- PA12) | Sửa đổi giấy chứng nhận tuân thủ dược điển châu Âu (CEP) của dược chất | x | x | x |
|
|
|
| Điều kiện cần đáp ứng (C) | Không có. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Giấy chứng nhận tuân thủ dược điển châu Âu (CEP) cho dược chất bản mới nhất còn hiệu lực kèm theo tất cả các phụ lục do EDQM (Hội đồng Châu Âu về chất lượng thuốc) cấp. 2. Kết quả phân tích lô của cơ sở sản xuất dược chất thể hiện chất lượng dược chất đáp ứng chuyên luận tương ứng của dược điển châu Âu đồng thời đáp ứng các chỉ tiêu chất lượng/ mức chất lượng bổ sung thêm được nêu trong CEP (nếu có). 3. Số liệu chứng minh cho bất kỳ thông số nào không được nêu trong CEP do cơ sở sản xuất dược chất tự công bố như dữ liệu về độ ổn định (S7) (trong trường hợp thời hạn phải kiểm tra lại chất lượng dược chất không nêu trong CEP) và các đặc tính lý hóa như kích thước phân tử, hiện tượng đa hình....của dược chất (nếu có). 4. Nếu thay đổi này là do thay đổi tiêu chuẩn chất lượng dược chất, bổ sung cam kết của cơ sở sản xuất thuốc thành phẩm về việc đã tiến hành và sẽ hoàn thành nghiên cứu độ ổn định của thuốc thành phẩm sản xuất từ dược chất đạt chất lượng theo tiêu chuẩn chất lượng dược chất mới; và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm đồng thời đề xuất biện pháp xử lý phù hợp. 5. Trong trường hợp tiêu chuẩn chất lượng đã được duyệt của dược chất không phải là tiêu chuẩn theo EP (do nhà sản xuất thành phẩm không áp dụng tiêu chuẩn chất lượng theo EP để kiểm tra chất lượng dược chất trước khi đưa vào sản xuất thành phẩm), nếu có thay đổi trong tiêu chuẩn chất lượng dược chất, yêu cầu nộp lại các phần S4-S5 theo ACTD. |
|
|
|
|
|
|
| Nội dung thay đổi 13 (MiV- PA13) | Bổ sung/Thay đổi cỡ lô đối với thuốc thành phẩm không vô khuẩn | x |
| x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Áp dụng cho thay đổi cỡ lô nhiều nhất đến 10 lần so với cỡ lô hiện tại. 2. Thay đổi này không ảnh hưởng đến tính ổn định của quy trình sản xuất. 3. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi. 4. Đối với thay đổi cỡ lô của thuốc thành phẩm vô khuẩn, xem MaV-7. Đối với thay đổi cỡ lô lớn hơn 10 lần so với cỡ lô đã đăng ký của thuốc thành phẩm không vô khuẩn, xem MaV-8. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Kế hoạch thẩm định quy trình và/hoặc báo cáo thẩm định quy trình sản xuất của ít nhất 03 lô sản xuất theo cỡ lô mới. 2. Bảng so sánh công thức lô sản xuất giữa cỡ lô hiện tại và cỡ lô mới. 3. Số liệu phân tích lô (dạng bảng so sánh) trên ít nhất 01 lô sản xuất giữa các thuốc thành phẩm trước và sau khi có thay đổi kèm theo bản cam kết của cơ sở sản xuất thuốc thành phẩm sẽ nộp dữ liệu phân tích lô của lô sản xuất tiếp theo. 4. Số liệu độ ổn định của thuốc thành phẩm sau khi có thay đổi và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đã được duyệt đồng thời đề xuất biện pháp xử lý phù hợp. 5. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. 6. Các phần từ P1- P3 theo ACTD đã cập nhật phù hợp với cỡ lô sản xuất mới. |
|
|
|
|
|
|
| Nội dung thay đổi 14 (MiV- PA14) | Giảm hoặc bỏ lượng đóng dư (overages) | x |
| x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Chỉ áp dụng cho thay đổi lượng đóng dư của dược chất. 2. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Giải trình về các lý do thay đổi. 2. Bảng so sánh công thức lô sản xuất đã được duyệt và công thức lô sản xuất mới. 3. Phiếu kiểm nghiệm của 02 lô thuốc thành phẩm sản xuất theo công thức lô mới. 4. Số liệu độ ổn định của thuốc thành phẩm sản xuất theo công thức lô mới và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. |
|
|
|
|
|
|
| Nội dung thay đổi 15 (MiV-PA15) | Thay đổi loại và/hoặc lượng tá dược a) Đối với dạng bào chế giải phóng ngay dùng đường uống (theo mức 1, Phần III (Thành phần và công thức)- hướng dẫn SUPAC-IR); b) Đối với dạng bào chế không đặc biệt khác như dung dịch uống, chế phẩm dùng ngoài. |
|
|
|
|
|
|
| TH1 | Các thuốc có chứng minh TĐSH | x | x | x |
|
| x |
| TH2 | Các thuốc không có chứng minh TĐSH | x | x | x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Các chỉ tiêu chất lượng và mức chất lượng trong tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi, trừ việc cập nhật thông tin về mô tả hình thức mới của thuốc (nếu có thay đổi hình thức thuốc). 2. Đối với các thuốc bào chế ở dạng rắn dùng đường uống: Số liệu so sánh biểu đồ hòa tan giữa các thuốc thành phẩm trước và sau khi có thay đổi đạt yêu cầu theo SUPAC-IR. 3. Thay thế một tá dược bằng một tá dược khác tương đương về đặc tính sử dụng (có cùng chức năng). 4. Đối với thay đổi về loại hoặc lượng tá dược của dạng bào chế giải phóng ngay dùng đường uống (theo level 2 và 3, phần III (Thành phần và công thức)- hướng dẫn SUPAC-IR), dạng bào chế giải phóng biến đổi dùng đường uống và các dạng bào chế đặc biệt khác, xem MaV-10. 5. Cơ sở sản xuất tá dược đáp ứng thực hành tốt sản xuất nguyên liệu làm thuốc (GMP) (thực hiện theo lộ trình quy định). |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Giải thích về đề nghị thay đổi bằng tài liệu phát triển dược học phù hợp. 2. Số liệu độ ổn định của thuốc thành phẩm sau khi có thay đổi và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 3. Đối với các dạng bào chế rắn dùng đường uống: Số liệu so sánh biểu đồ hòa tan trên ít nhất 01 lô (lô pilot hoặc lô sản xuất) đại diện giữa thuốc thành phẩm trước và sau khi có thay đổi. 4. Giải trình các căn cứ khoa học của việc không nộp số liệu nghiên cứu tương đương sinh học của thuốc thành phẩm sau khi có thay đổi (đối với các thuốc đã được cấp số đăng ký là thuốc có chứng minh tương đương sinh học). 5. Bảng so sánh công thức bào chế đã được duyệt và công thức bào chế mới (có in đậm các thay đổi), trong đó lượng mỗi thành phần được thể hiện dưới dạng phần trăm khối lượng trên tổng khối lượng các thành phần trong công thức. 6. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành của thuốc thành phẩm sau khi có thay đổi trong đó đã cập nhật các thay đổi về mô tả hình thức mới của thuốc (trong trường hợp có thay đổi trong mô tả hình thức thuốc). 7. Số liệu phân tích lô (dạng bảng so sánh) trên ít nhất 02 lô sản xuất (hoặc 01 lô sản xuất và 02 lô pilot) giữa các thuốc thành phẩm trước và sau khi có thay đổi. 8. Tiêu chuẩn chất lượng của tá dược mới (nếu có sử dụng thêm tá dược chưa có trong công thức bào chế đã được duyệt). 9. Đối với tá dược có nguồn gốc từ động vật nhai lại, yêu cầu nộp giấy chứng nhận không có TSE hoặc BSE do cơ quan quản lý thú y có thẩm quyền cấp. 10. Công thức lô sản xuất mới. 11. Kế hoạch thẩm định và/hoặc báo cáo thẩm định quy trình sản xuất của ít nhất 03 lô thuốc thành phẩm sản xuất theo công thức bào chế mới. 12. Các phần P3.1 đến P3.4 theo ACTD đã cập nhật phù hợp với công thức bào chế mới. 13. Giải trình phù hợp kèm theo số liệu thực nghiệm chứng minh (nếu cần) về việc các thay đổi này không làm ảnh hưởng đến kết quả của các quy trình phân tích trong tiêu chuẩn chất lượng xuất xưởng/ tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. Nếu thay đổi này dẫn đến việc phải thay đổi quy trình phân tích trong tiêu chuẩn chất lượng xuất xưởng/ tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm, yêu cầu tham chiếu MiV-PA27 để bổ sung đầy đủ các tài liệu theo quy định. 14. Giấy tờ pháp lý của cơ sở sản xuất tá dược mới theo quy định tại khoản 11 Điều 22 Thông tư này. Không yêu cầu tài liệu này đối với tá dược đã có Giấy đăng ký lưu hành tại Việt Nam. |
|
|
|
|
|
|
| Nội dung thay đổi 16 (MiV-PA16) | Thay đổi khối lượng màng bao viên hoặc khối lượng và/hoặc kích cỡ vỏ nang đối với dạng bào chế rắn giải phóng ngay dùng đường uống |
|
|
|
|
|
|
| TH1 | Các thuốc có chứng minh TĐSH | x | x | x |
|
| x |
| TH2 | Các thuốc không có chứng minh TĐSH | x | x | x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Đối với các thuốc bào chế ở dạng rắn dùng đường uống: Số liệu so sánh biểu đồ hòa tan giữa các thuốc thành phẩm trước và sau khi có thay đổi đạt yêu cầu theo SUPAC-IR. 2. Các chỉ tiêu chất lượng và mức chất lượng trong tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi, trừ việc cập nhật thông tin về mô tả hình thức mới của thuốc (nếu có thay đổi hình thức thuốc). 3. Thay đổi khối lượng màng bao viên hoặc khối lượng và/hoặc kích cỡ vỏ nang đối với dạng bào chế rắn giải phóng biến đổi dùng đường uống, xem MaV-11. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Số liệu so sánh biểu đồ hòa tan trên ít nhất 01 lô (lô pilot hoặc lô sản xuất) giữa các thuốc thành phẩm trước và sau khi có thay đổi. 2. Giải trình các căn cứ khoa học của việc không nộp số liệu nghiên cứu tương đương sinh học của thuốc thành phẩm sau khi có thay đổi (đối với các thuốc đã được cấp số đăng ký là thuốc có chứng minh tương đương sinh học). 3. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành của thuốc thành phẩm sau khi có thay đổi trong đó đã cập nhật các thay đổi về mô tả hình thức mới của thuốc (trong trường hợp có thay đổi mô tả hình thức thuốc). 4. Cam kết của cơ sở sản xuất thuốc thành phẩm và cơ sở đăng ký về việc thay đổi này không ảnh hưởng đến quy trình phân tích trong tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. 5. Bảng so sánh công thức bào chế cho 01 đơn vị liều và công thức lô sản xuất của các thuốc thành phẩm trước và sau khi có thay đổi. 6. Nếu thay đổi khối lượng màng bao viên: Số liệu nghiên cứu độ ổn định của thuốc thành phẩm sau khi có thay đổi và báo cáo nếu có bất cứ kết quả nào không đạt so với tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý. Nếu thay đổi khối lượng và/ hoặc kích cỡ vỏ nang: cam kết của cơ sở sản xuất thuốc thành phẩm về việc đã bắt đầu và sẽ hoàn thành việc nghiên cứu độ ổn định của thuốc thành phẩm sau khi có thay đổi. 7. Phần P3.1 đến P3.4 theo ACTD đã cập nhật các thay đổi tương ứng. |
|
|
|
|
|
|
| Nội dung thay đổi 17 (MiV- PA17) | Thay đổi chất tạo màu/tạo mùi của thuốc [thêm vào, bỏ bớt hoặc thay thế (các) chất tạo màu/tạo mùi] | x | x | x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Các tá dược thay đổi có cùng chức năng. Sự thay đổi này không làm thay đổi đặc tính hòa tan của thuốc thành phẩm đối với các dạng bào chế rắn dùng đường uống. 2. Chất tạo màu/tạo mùi mới không bị cấm dùng trong dược phẩm. 3. Các chỉ tiêu chất lượng và mức chất lượng trong tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi trừ việc cập nhật mô tả về màu sắc và/ hoặc mùi vị mới của thuốc. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Bảng so sánh các thông tin liên quan đến tính chất và khối lượng sử dụng giữa chất tạo màu/ tạo mùi mới và chất tạo màu/ tạo mùi đang sử dụng . 2. Công thức bào chế cho 01 đơn vị liều và công thức lô sản xuất thuốc thành phẩm sau khi có thay đổi. 3. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành của thuốc thành phẩm sau khi có thay đổi đã cập nhật các mô tả về màu sắc và/ hoặc mùi vị mới của thuốc. 4. Số liệu độ ổn định của thuốc thành phẩm sau khi có thay đổi và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 5. Đối với tá dược có nguồn gốc từ động vật nhai lại, yêu cầu nộp giấy chứng nhận không có TSE hoặc BSE do cơ quan quản lý thú y có thẩm quyền cấp. 6. Giải trình phù hợp kèm theo số liệu thực nghiệm chứng minh (nếu cần) về việc các thay đổi này không làm ảnh hưởng đến kết quả các quy trình phân tích trong tiêu chuẩn chất lượng xuất xưởng/ tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. Nếu thay đổi này dẫn đến việc phải thay đổi quy trình phân tích trong tiêu chuẩn chất lượng thành phẩm đã được duyệt, yêu cầu tham chiếu MiV-PA27 để bổ sung các tài liệu theo quy định. 7. Phần P3.1 đến P3.4 theo ACTD đã được cập nhật các thay đổi phù hợp. |
|
|
|
|
|
|
| Nội dung thay đổi 18 (MiV-PA18) | Bỏ bớt dung môi/dung dịch hòa tan, phân tán, pha loãng thuốc | x |
| x | x |
|
|
| Điều kiện cần đáp ứng (C) | 1. Thay đổi này không dẫn đến bất kỳ thay đổi nào về dạng bào chế, phác đồ điều trị, chỉ định, cách dùng của thuốc. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Giải trình về việc bỏ bớt dung môi, dung dịch hòa tan/ phân tán/ pha loãng của thuốc và trình bày về các biện pháp thay thế khác để có dung môi, dung dịch hòa tan/ phân tán/ pha loãng. |
|
|
|
|
|
|
| Nội dung thay đổi 19 (MiV- PA19) | Thay đổi trong kiểm soát trong quy trình sản xuất đối với thuốc thành phẩm (bao gồm theo hướng chặt chẽ hơn và bổ sung phép thử mới) | x | x | x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi. 2. Thay đổi này không phải là hệ quả của bất kỳ cam kết nào về việc xem xét lại giới hạn trong tiêu chuẩn chất lượng từ lần thẩm định trước. 3. Thay đổi này không phải là biến cố ngoại ý phát sinh trong quá trình sản xuất như: xuất hiện tạp chất mới chưa xác định, thay đổi giới hạn tổng tạp chất... 4. Bất kỳ quy trình phân tích mới nào đều không liên quan đến kỹ thuật mới không theo chuẩn hoặc một kỹ thuật cơ bản được sử dụng theo cách mới. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Mô tả quy trình phân tích và tóm tắt số liệu thẩm định quy trình phân tích đối với các quy trình phân tích mới (nếu có sử dụng quy trình phân tích mới). 2. Các chỉ tiêu kiểm soát trong quy trình sản xuất sau khi có thay đổi kèm theo các giải trình và số liệu thẩm định quy trình phù hợp. 3. So sánh số liệu phân tích lô trên ít nhất 02 lô (lô pilot hoặc lô sản xuất) của thuốc thành phẩm sau khi có thay đổi. 4. Bảng so sánh kiểm soát trong quy trình sản xuất trước và sau khi có thay đổi |
|
|
|
|
|
|
| Nội dung thay đổi 20 (MiV- PA20) | Thay đổi nhỏ trong quy trình sản xuất thuốc thành phẩm không vô khuẩn | x |
| x |
|
| x |
| Điều kiện cần đáp ứng (C) | 1. Địa điểm sản xuất không thay đổi. 2. Nguyên tắc sản xuất chung không thay đổi. 3. Thay đổi này không ảnh hưởng tiêu cực đến chất lượng, an toàn và hiệu quả của thuốc thành phẩm. 4. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi. 5. Đối với các thuốc thành phẩm bào chế ở dạng rắn dùng đường uống: Số liệu so sánh biểu đồ hòa tan giữa các thuốc thành phẩm trước và sau khi thay đổi đạt yêu cầu theo quy định tại SUPAC-IR và SUPAC-MR. 6. Đối với thay đổi lớn trong quy trình sản xuất thuốc thành phẩm, áp dụng MaV-9. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Mô tả quy trình sản xuất mới và giải trình về mặt kỹ thuật cho sự thay đổi này. 2. Đối với thuốc dạng bán rắn và dạng hỗn dịch: Kế hoạch thẩm định và/ hoặc báo cáo thẩm định quy trình sản xuất. 3. Đối với các dạng bào chế rắn dùng đường uống: Số liệu so sánh biểu đồ hòa tan trên ít nhất 01 lô sản xuất đại diện giữa các thuốc thành phẩm trước và sau khi có thay đổi. 4. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. 5. Giải trình các căn cứ khoa học của việc không nộp số liệu nghiên cứu tương đương sinh học của thuốc thành phẩm sau khi có thay đổi (đối với các thuốc đã được cấp số đăng ký là thuốc có chứng minh tương đương sinh học). 6. Số liệu phân tích lô thành phẩm (dạng bảng so sánh) trên ít nhất 01 lô sản xuất giữa thuốc thành phẩm trước và sau khi có thay đổi; số liệu phân tích lô cho 02 lô sản xuất đầy đủ kế tiếp phải sẵn có để nộp khi có yêu cầu. 7. Cam kết của cơ sở sản xuất thuốc thành phẩm và cơ sở đăng ký về việc đã bắt đầu tiến hành và sẽ hoàn thành nghiên cứu độ ổn định của thuốc thành phẩm sau khi thay đổi và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 8. Bảng so sánh quy trình sản xuất mới và quy trình sản xuất đã được duyệt có in đậm những nội dung thay đổi. |
|
|
|
|
|
|
| Nội dung thay đổi 21 (MiV- PA21) | Thay đổi tiêu chuẩn chất lượng của tá dược áp dụng đối với các trường hợp sau: a) Giới hạn chỉ tiêu chất lượng chặt chẽ hơn; b) Bổ sung chỉ tiêu chất lượng và mức chất lượng mới. | x | x |
|
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Áp dụng cho các trường hợp tiêu chuẩn chất lượng đã được duyệt của tá dược không theo dược điển. Nếu tiêu chuẩn chất lượng đã được duyệt của tá dược theo dược điển, xem MiV-N6. 2. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi. 3. Thay đổi này không phải là kết quả của biến cố ngoại ý xuất hiện trong quá trình sản xuất hoặc do quan ngại về độ ổn định. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Bảng so sánh tiêu chuẩn chất lượng tá dược trước và sau khi có thay đổi có in đậm những nội dung thay đổi. 2. Mô tả quy trình phân tích mới (trong trường hợp bổ sung chỉ tiêu chất lượng mới). Phiếu kiểm nghiệm tá dược trong trường hợp bổ sung chỉ tiêu chất lượng và mức chất lượng mới. |
|
|
|
|
|
|
| Nội dung thay đổi 22 (MiV- PA22) | Thay đổi quy trình phân tích của tá dược, bao gồm thay thế quy trình phân tích đã được duyệt bằng quy trình phân tích mới. | x | x |
|
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Đã có số liệu thẩm định quy trình phân tích mới đạt yêu cầu. 2. Kết quả thẩm định quy trình phân tích mới cho thấy quy trình phân tích mới ít nhất là tương đương với quy trình phân tích đã được duyệt. 3. Không có sự thay đổi về giới hạn tổng tạp chất. 4. Chỉ áp dụng đối với các chỉ tiêu chất lượng đã được duyệt trong tiêu chuẩn. 5. Không có sự xuất hiện của các tạp chất mới chưa xác định. Áp dụng cho các trường hợp tiêu chuẩn chất lượng đã được duyệt của tá dược không theo dược điển. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Mô tả quy trình phân tích mới kèm theo bảng so sánh các thay đổi giữa quy trình phân tích mới và quy trình phân tích đã được duyệt. 2. Đối với các quy trình định lượng (bao gồm cả định lượng tạp chất): Số liệu so sánh kết quả thẩm định quy trình phân tích chứng minh quy trình phân tích mới và quy trình phân tích đã được duyệt là tương đương. |
|
|
|
|
|
|
| Nội dung thay đổi 23 (MiV- PA23) | Thay đổi nguồn gốc vỏ nang cứng, áp dụng đối với các trường hợp sau: a) Thay đổi nguồn gốc nguyên liệu sản xuất vỏ nang cứng; b) Thay đổi/bổ sung cơ sở sản xuất vỏ nang cứng. | x | x | x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Thay đổi từ vỏ nang dùng nguyên liệu có nguồn gốc động vật có nguy cơ gây bệnh TSE sang vỏ nang dùng nguyên liệu có nguồn gốc thực vật hoặc vỏ nang tổng hợp và ngược lại. 2. Không thay đổi công thức bào chế và quy trình sản xuất đã được duyệt của thuốc thành phẩm. 3. Không áp dụng khi thay đổi từ vỏ nang cứng sang vỏ nang mềm. 4. Tiêu chuẩn chất lượng đã được duyệt của các tá dược (trừ tiêu chuẩn chất lượng vỏ nang mới), tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi. 5. Cơ sở sản xuất vỏ nang đáp ứng thực hành tốt sản xuất (GMP) (thực hiện theo lộ trình quy định). |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Số liệu so sánh biểu đồ hòa tan trên 01 lô (lô pilot hoặc lô sản xuất) đại diện giữa các thuốc thành phẩm trước và sau khi có thay đổi. 2. Thành phần và tiêu chuẩn chất lượng của vỏ nang cứng mới. 3. Phiếu kiểm nghiệm vỏ nang cứng mới. 4. Số liệu độ ổn định của thuốc thành phẩm sau khi có thay đổi và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 5. Đối với vỏ nang cứng sản xuất từ nguyên liệu có nguồn gốc từ động vật nhai lại, yêu cầu nộp giấy chứng nhận không có TSE hoặc BSE do cơ quan quản lý thú ý có thẩm quyền cấp. 6. Cam kết của cơ sở sản xuất thuốc về việc nguyên liệu sản xuất vỏ nang cứng là tinh khiết từ các nguồn tổng hợp hoặc động vật hoặc thực vật. 7. Số liệu chứng minh vỏ nang mới không làm ảnh hưởng đến kết quả thử độ hòa tan theo quy trình phân tích trong tiêu chuẩn chất lượng thuốc thành phẩm đã được duyệt (nếu trong tiêu chuẩn chất lượng thuốc thành phẩm có chỉ tiêu chất lượng này). 8. Giấy tờ pháp lý của cơ sở sản xuất vỏ nang chứng minh cơ sở đáp ứng thực hành tốt sản xuất nguyên liệu làm thuốc (GMP) (thực hiện theo lộ trình quy định). Không yêu cầu tài liệu này đối với vỏ nang đã có Giấy đăng ký lưu hành tại Việt Nam. |
|
|
|
|
|
|
| Nội dung thay đổi 24 (MiV- PA24) | Thay đổi trong tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành của thuốc thành phẩm, áp dụng đối với các trường hợp sau: a) Giới hạn chỉ tiêu chất lượng chặt chẽ hơn; b) Bổ sung chỉ tiêu chất lượng và mức chất lượng. | x | x | x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Áp dụng trong trường hợp tiêu chuẩn chất lượng đã được duyệt của thuốc thành phẩm không theo một trong các dược điển tham chiếu. 2. Thay đổi này không phải là kết quả của biến cố ngoại ý phát sinh trong quá trình sản xuất hoặc do quan ngại về độ ổn định. 3. Quy trình phân tích không thay đổi hoặc thay đổi rất ít (không cần thiết phải tiến hành thẩm định lại quy trình). 4. Nếu có thay đổi quy trình phân tích, áp dụng thêm MiV-PA27. Nếu mở rộng giới hạn chỉ tiêu chất lượng, bỏ bớt chỉ tiêu chất lượng và mức chất lượng, xem MaV-6. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | (a) Giới hạn chỉ tiêu chất lượng chặt chẽ hơn: 1. Bảng so sánh tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành trước và sau khi có thay đổi của thuốc thành phẩm có in đậm những nội dung thay đổi. 2. Phiếu kiểm nghiệm hoặc so sánh số liệu phân tích lô của tất cả các chỉ tiêu chất lượng trong tiêu chuẩn chất lượng mới trên ít nhất 02 lô sản xuất của thuốc thành phẩm. 3. Giải trình về mặt kỹ thuật các lý do dẫn đến đề nghị thay đổi này. (b) Bổ sung chỉ tiêu chất lượng và mức chất lượng: Ngoài các tài liệu nêu trên yêu cầu: 4. Mô tả quy trình phân tích các chỉ tiêu chất lượng mới của thuốc thành phẩm và cung cấp số liệu thẩm định các quy trình phân tích của các chỉ tiêu chất lượng mới này (đối với các quy trình phân tích không có trong dược điển, quy trình phân tích trong dược điển mới khác quy trình phân tích dược điển cũ). 5. Số liệu độ ổn định của thuốc thành phẩm sau khi có thay đổi và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành mới đồng thời đề xuất biện pháp xử lý phù hợp. |
|
|
|
|
|
|
| Nội dung thay đổi 25 (MiV- PA25) | Thay đổi vết khắc, hình ảnh hoặc các ký hiệu khác trên viên nén hoặc hình ảnh/ ký hiệu in trên viên nén, viên nang bao gồm thay đổi/ bổ sung mực in dùng để in lên thuốc thành phẩm | x | x |
|
|
|
|
| Điều kiện cần đáp ứng (C) | (a) Tất cả các thay đổi tại mục này, trừ đường kẻ/vạch bẻ thuốc: 1. Các ký hiệu mới không gây nhầm lẫn với thuốc thành phẩm khác đã được đăng ký. 2. Loại mực mới dự kiến sử dụng không được nằm trong danh mục các chất cấm dùng và phải đáp ứng các tiêu chuẩn để dùng trong thực phẩm hoặc dược phẩm. 3. Tiêu chuẩn chất lượng xuất xưởng và Tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi ngoại trừ thay đổi về mô tả hình thức viên. (b) Thay đổi đường kẻ/vạch bẻ thuốc Ngoài những điều kiện có liên quan nêu trên yêu cầu: 4. Đường kẻ/vạch bẻ thuốc không nhằm mục đích làm đẹp. 5. Áp dụng đối với bổ sung hoặc bỏ đường kẻ/vạch bẻ thuốc. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | (a) Tất cả các thay đổi tại mục này, trừ đường kẻ/vạch bẻ thuốc: 1. Thành phần và tiêu chuẩn chất lượng của mực in mới dự kiến sử dụng. 2. Phiếu kiểm nghiệm của mực in mới/ nguyên liệu dùng để in mới (loại dùng trong thực phẩm, dược phẩm). 3. Mô tả chi tiết (bằng hình ảnh hoặc bản viết) các vết khắc/hình ảnh/ký hiệu trên thuốc thành phẩm trước và sau khi thay đổi. 4. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành của thuốc thành phẩm đã cập nhật mô tả mới về hình thức viên. (b) Thay đổi đường kẻ/vạch bẻ thuốc: Ngoài những tài liệu có liên quan nêu trên yêu cầu: 5. Giải trình về lý do thay đổi (ví dụ do thay đổi liều dùng). 6. Phiếu kiểm nghiệm cho 02 lô (lô sản xuất hoặc lô pilot) của thuốc thành phẩm sau khi có thay đổi. 7. Số liệu kiểm tra độ đồng đều đơn vị liều giữa các phần của viên thuốc sau khi bẻ tại thời điểm xuất xưởng (trong trường hợp bổ sung đường kẻ/ vạch bẻ thuốc). |
|
|
|
|
|
|
| Nội dung thay đổi 26 (MiV- PA26) | Thay đổi kích thước và/hoặc hình dạng viên nén, viên nang, viên đạn hoặc viên đặt âm đạo mà không thay đổi loại và lượng của các thành phần trong viên và khối lượng trung bình viên đối với: a) Dạng bào chế rắn giải phóng ngay dùng đường uống, viên đạn và viên đặt âm đạo; b) Các trường hợp không phải dạng bào chế rắn giải phóng ngay dùng đường uống, viên đạn và viên đặt âm đạo. | x | x | x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Có số liệu so sánh biểu đồ hòa tan của thuốc thành phẩm trước và sau khi có thay đổi (đối với các trường hợp có áp dụng) đạt quy định theo SUPAC-IR và SUPAC-MR. 2. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi ngoại trừ thay đổi trong mô tả kích thước và/hoặc hình dạng thuốc thành phẩm. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | (a) Dạng bào chế rắn giải phóng ngay dùng đường uống, viên đạn, viên đặt âm đạo: 1. Mô tả chi tiết bằng hình vẽ hoặc bản viết hình dạng/ kích thước thuốc thành phẩm trước và sau khi có thay đổi. 2. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành của thuốc thành phẩm đã cập nhật mô tả mới về hình dạng/ kích thước của thuốc thành phẩm. 3. Số liệu so sánh biểu đồ hòa tan trên ít nhất 01 lô (lô pilot hoặc lô sản xuất) giữa các thuốc thành phẩm trước và sau khi có thay đổi. 4. Số liệu kiểm tra độ đồng đều đơn vị liều giữa các phần viên thuốc sau khi bẻ tại thời điểm xuất xưởng đáp ứng các yêu cầu của dược điển (chỉ áp dụng đối với thuốc thành phẩm có đường kẻ/vạch bẻ thuốc). (b) Không phải dạng bào chế rắn giải phóng ngay dùng đường uống, viên đạn, viên đặt âm đạo: Ngoài những tài liệu nêu trên yêu cầu: 5. Giải trình các căn cứ khoa học của việc không nộp số liệu nghiên cứu tương đương sinh học của thuốc thành phẩm sau khi có thay đổi (đối với các thuốc đã được cấp số đăng ký là thuốc có chứng minh tương đương sinh học). |
|
|
|
|
|
|
| Nội dung thay đổi 27 (MiV- PA27) | Thay đổi quy trình phân tích trong tiêu chuẩn chất lượng thuốc thành phẩm (bao gồm thay thế hoặc bổ sung quy trình phân tích) | x | x |
|
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Các chỉ tiêu chất lượng và mức chất lượng trong tiêu chuẩn chất lượng đã được duyệt của thuốc thành phẩm không bị ảnh hưởng ngoại trừ theo hướng chặt chẽ hơn. 2. Kết quả thẩm định quy trình phân tích cho thấy quy trình phân tích mới ít nhất là tương đương với quy trình phân tích đã được duyệt. Thay đổi này không phải là kết quả của biến cố ngoại ý phát sinh trong quá trình sản xuất hoặc do quan ngại về độ ổn định. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Mô tả quy trình phân tích mới. 2. Số liệu thẩm định/ thẩm tra quy trình phân tích mới và bảng so sánh các kết quả phân tích thu được từ quy trình phân tích đã được duyệt và quy trình phân tích mới. 3. Phiếu kiểm nghiệm của 02 lô sản xuất của thuốc thành phẩm được kiểm tra theo tiêu chuẩn chất lượng thuốc thành phẩm đã thay đổi. 4. Giải trình lý do thay đổi. Bảng so sánh tiêu chuẩn chất lượng xuất xưởng/ tiêu chuẩn chất lượng lưu hành trước và sau khi có thay đổi của thuốc thành phẩm. |
|
|
|
|
|
|
| Nội dung thay đổi 28 (MiV- PA28) | Thay đổi liên quan đến bao bì đóng gói sơ cấp của thuốc thành phẩm không vô khuẩn gồm: a) Thay đổi thành phần và lượng các thành phần của chất liệu bao bì; b) Thay đổi loại bao bì và/ hoặc chất liệu bao bì. | x |
| x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi. 2. Đối với thay đổi nguyên liệu bao bì sơ cấp của thuốc thành phẩm vô khuẩn, xem MaV-12. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Giải thích lý do thay đổi chất liệu bao bì đóng gói sơ cấp và các nghiên cứu khoa học phù hợp liên quan đến bao bì đóng gói sơ cấp mới. 2. Đối với dạng bào chế bán rắn và lỏng, cung cấp các bằng chứng chứng minh không có sự tương tác giữa thuốc và chất liệu bao bì đóng gói mới (Thí dụ: các thành phần của chất liệu bao bì không đi vào thuốc và thành phần của thuốc không bị mất vào bao bì). 3. Bảng so sánh tiêu chuẩn chất lượng giữa chất liệu bao bì sơ cấp mới và chất liệu bao bì sơ cấp đã được duyệt. 4. Số liệu độ ổn định của thuốc thành phẩm trong bao bì đóng gói sơ cấp mới và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 5. Mẫu nhãn đã được duyệt và mẫu nhãn mới (nếu có thay đổi). |
|
|
|
|
|
|
| Nội dung thay đổi 29 (MiV-PA29) | Thay đổi cơ sở đóng gói thứ cấp | x |
|
|
|
|
|
| Điều kiện cần đáp ứng (C) | Chỉ thay đổi cơ sở đóng gói thứ cấp, tất cả các yếu tố khác không thay đổi |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Giấy chứng nhận GMP hoặc CPP đối với cơ sở đóng gói tại nước ngoài và Giấy chứng nhận đủ điều kiện kinh doanh đối với cơ sở đóng gói trong nước. Thư của chủ sở hữu sản phẩm cho phép cơ sở đóng gói mới thực hiện việc đóng gói thứ cấp (nếu có). |
|
|
|
|
|
|
| Nội dung thay đổi 30 (MiV- PA30) | Thay đổi hoặc bổ sung quy cách đóng gói thuốc thành phẩm trong bao bì sơ cấp và/hoặc thay đổi hình dạng hoặc kích thước đồ bao gói sơ cấp hoặc hệ thống đóng kín đối với thuốc thành phẩm không vô khuẩn. |
|
|
|
|
|
|
| TH1 | Có thay đổi chất liệu bao bì đóng gói: Phối hợp thêm với nội dung MiV-PA28 | x |
| x |
|
|
|
| TH2 | Không thay đổi chất liệu/tiêu chuẩn bao bì đóng gói | x |
|
|
|
|
|
| TH3 | Có thay đổi chỉ tiêu thể tích hoặc khối lượng đóng gói. | x | x | x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Tiêu chuẩn chất lượng xuất xưởng và tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm không thay đổi. 2. Quy cách đóng gói mới phù hợp với liều dùng và thời gian sử dụng đã được duyệt trong hướng dẫn sử dụng. 3. Thay đổi hoặc bổ sung quy cách đóng gói thuốc thành phẩm trong bao bì sơ cấp và/hoặc thay đổi hình dạng hoặc kích thước đồ bao gói sơ cấp hoặc hệ thống đóng kín của thuốc vô khuẩn dạng rắn và dạng dung dịch, xem MaV-13. 4. Chất liệu bao bì đóng gói không thay đổi. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Giải thích lý do thay đổi quy cách đóng gói. 2. Mẫu nhãn đã được duyệt và mẫu nhãn mới (nếu có thay đổi). 3. Các phần hồ sơ P3, P7 đã sửa đổi các thông tin liên quan (nếu có thay đổi) 4. Cam kết của cơ sở sản xuất thuốc thành phẩm và cơ sở đăng ký về việc đã bắt đầu tiến hành và sẽ hoàn thành nghiên cứu độ ổn định của thuốc thành phẩm trong bao bì đóng gói sơ cấp mới và báo cáo nếu có bất kỳ kết quả nào không đạt tiêu chuẩn chất lượng lưu hành đồng thời đề xuất biện pháp xử lý phù hợp. 5. Bổ sung tiêu chuẩn chất lượng thành phẩm cập nhật và phiếu kiểm nghiệm đối với quy cách đóng gói mới trong trường hợp có thay đổi chỉ tiêu thể tích hoặc khối lượng đóng gói. |
|
|
|
|
|
|
| Nội dung thay đổi 31 (MiV- PA31) | Thay đổi, bổ sung quy cách đóng gói thứ cấp của thuốc thành phẩm | x |
|
|
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Quy cách đóng gói đề nghị thay đổi, bổ sung phù hợp với liều dùng đã được duyệt trong hướng dẫn sử dụng. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Hướng dẫn sử dụng đã được duyệt. 2. Mẫu nhãn đã được duyệt và mẫu nhãn mới (nếu có thay đổi). 3. Giải trình về việc thay đổi, bổ sung quy cách đóng gói thứ cấp là phù hợp với liều dùng đã được duyệt trong hướng dẫn sử dụng. |
|
|
|
|
|
|
| Nội dung thay đổi 32 (MiV- PA32) | Các thay đổi của bao bì (sơ cấp) không tiếp xúc trực tiếp với thuốc thành phẩm như màu của nắp bật, vạch màu trên ống, thay đổi chụp bảo vệ kim tiêm (sử dụng nhựa khác) | x |
| x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Thay đổi này không liên quan đến phần nguyên liệu bao bì có ảnh hưởng đến sự phân liều, sự sử dụng, tính an toàn hoặc độ ổn định của thuốc. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Các phần hồ sơ liên quan đến thay đổi (trình bày theo quy định của ACTD). 2. Bảng so sánh các nội dung thay đổi. Mẫu nhãn đã được duyệt và mẫu nhãn mới (nếu có thay đổi). |
|
|
|
|
|
|
| Nội dung thay đổi 33 (MiV- PA33) | Bổ sung hoặc thay thế dụng cụ đo lường của các dạng thuốc lỏng dùng đường uống và các dạng bào chế khác | x |
| x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Kích cỡ và nếu cần, mức độ chính xác của dụng cụ đo lường mới phải phù hợp với liều dùng đã được duyệt của thuốc thành phẩm. 2. Dụng cụ mới phải tương thích với thuốc thành phẩm. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Mô tả dụng cụ đo lường mới (bao gồm hình vẽ, nếu có). 2. Thông tin về thành phần nguyên liệu dùng trong sản xuất dụng cụ đo lường. Nguyên liệu dùng trong sản xuất dụng cụ đo lường phải đáp ứng tiêu chuẩn chất lượng của dược điển tham chiếu (nếu có tiêu chuẩn của nguyên liệu này trong dược điển). 3. Thuyết minh về sự phù hợp của kích cỡ và mức độ chính xác của dụng cụ đo lường mới với liều dùng đã được duyệt của thuốc thành phẩm. |
|
|
|
|
|
|
| Nội dung thay đổi 34 (MiV- PA34) | Giảm hạn dùng của thuốc thành phẩm a) Đóng gói trong bao bì sơ cấp theo các quy cách đóng gói đã được duyệt và/ hoặc b) Sau khi mở nắp lần đầu và/hoặc c) Sau khi hòa tan, phân tán hoặc pha loãng | x |
| x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Đối với (a) & (b) - Các nghiên cứu phải thể hiện được sự phù hợp với tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. 2. Đối với (c) - Các nghiên cứu phải thể hiện được sự phù hợp với tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm sau khi hòa tan, phân tán hoặc pha loãng. 3. Trường hợp tăng hạn dùng thuốc thành phẩm, xem MaV-15. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Số liệu độ ổn định dài hạn phù hợp với hạn dùng mới trên ít nhất 02 lô (lô pilot hoặc lô sản xuất) kèm theo các kết quả thử nghiệm vi sinh vật phù hợp của thuốc thành phẩm trong các trường hợp: a) Đóng gói trong bao bì sơ cấp theo các quy cách đóng gói đã được duyệt và/hoặc b) Sau khi mở nắp lần đầu và/hoặc c) Sau khi hòa tan, pha loãng hoặc phân tán 2. Giải trình các lý do đề nghị giảm hạn dùng của thuốc. 3. Báo cáo số lượng thuốc đang lưu hành trên thị trường. 4. Tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. |
|
|
|
|
|
|
| Nội dung thay đổi 35 (MiV- PA35) | Thay đổi điều kiện bảo quản của thuốc thành phẩm (khắc nghiệt hơn so với điều kiện bảo quản đã được chấp thuận) a) Trong bao bì đóng gói sơ cấp theo các quy cách đóng gói đã được duyệt và/hoặc b) Sau khi mở nắp lần đầu và/hoặc c) Sau khi hòa tan, phân tán hoặc pha loãng. | x |
| x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Đối với (a) & (b) - Các nghiên cứu phải thể hiện được sự phù hợp với tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm. 2. Đối với (c) - Các nghiên cứu phải thể hiện được sự phù hợp với tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm sau khi hòa tan, phân tán hoặc pha loãng. 3. Nếu thay đổi điều kiện bảo quản (ít khắc nghiệt hơn so với điều kiện bảo quản đã được duyệt), xem MaV-16. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Số liệu độ ổn định dài hạn của thuốc thành phẩm ở điều kiện bảo quản mới trong khoảng thời gian phù hợp với hạn dùng tương ứng đã được duyệt trên ít nhất 02 lô (lô pilot hoặc lô sản xuất) kèm theo các kết quả thử nghiệm vi sinh vật phù hợp trong các trường hợp: a) Khi đóng gói trong bao bì sơ cấp theo các quy cách đóng gói đã được duyệt và/hoặc b) Sau khi mở nắp lần đầu và/ hoặc c) Sau khi hòa tan, phân tán hoặc pha loãng. 2. Giải thích về mặt kỹ thuật các lý do thay đổi điều kiện bảo quản. 3. Tiêu chuẩn chất lượng lưu hành đã được duyệt của thuốc thành phẩm |
|
|
|
|
|
|
| Nội dung thay đổi 36 (MiV- PA36) | Thay đổi cơ sở đăng ký | x |
|
|
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Áp dụng đối với thay đổi cơ sở đăng ký |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Giấy chứng nhận đủ điều kiện kinh doanh có tên, địa chỉ mới hoặc của cơ sở đăng ký mới (nếu là cơ sở đăng ký của Việt Nam). 2. Giấy phép sản xuất, kinh doanh thuốc do cơ quan quản lý nhà nước có thẩm quyền nước ngoài cấp và Giấy phép thành lập Văn phòng đại diện tại Việt Nam với tên, địa chỉ mới hoặc của cơ sở đăng ký mới (nếu là cơ sở đăng ký của nước ngoài). |
|
|
|
|
|
|
| Nội dung thay đổi 37 (MiV- PA37) | Công bố thuốc có chứng minh tương đương sinh học khi: 1. Trường hợp 1: Các thuốc thuộc trường hợp không bắt buộc phải nộp báo cáo tương đương sinh học trong hồ sơ đăng ký cấp giấy đăng ký lưu hành sau khi được cấp giấy đăng ký lưu hành có đề nghị công bố thuốc có chứng minh tương đương sinh học; 2. Trường hợp 2: Các thuốc đã được Cục Quản lý Dược công bố tương đương sinh học, khi cấp lại giấy đăng ký lưu hành dẫn đến thay đổi số giấy đăng ký lưu hành, có hoặc không kèm theo các thay đổi khác không ảnh hưởng/ảnh hưởng không đáng kể đến sinh khả dụng của thuốc. 3. Trường hợp 3: Các thuốc đã được Cục Quản lý Dược công bố tương đương sinh học, khi cấp lại giấy đăng ký lưu hành dẫn đến thay đổi số giấy đăng ký lưu hành và có các thay đổi khác ảnh hưởng đáng kể đến sinh khả dụng của thuốc. | x |
|
|
|
| x |
| Điều kiện cần đáp ứng (C) | 1. Trường hợp 1: Có báo cáo nghiên cứu chứng minh tương đương sinh học đáp ứng quy định. 2. Trường hợp 2: Thuốc được cấp lại giấy đăng ký lưu hành không có bất kỳ thay đổi nào so với thuốc đã được công bố có chứng minh tương đương sinh học hoặc nếu có các thay đổi, bổ sung đã được chứng minh không ảnh hưởng/ảnh hưởng không đáng kể đến sinh khả dụng của thuốc. 3. Trường hợp 3: Thuốc được cấp lại giấy đăng ký lưu hành có các thay đổi, bổ sung ảnh hưởng đáng kể đến sinh khả dụng của thuốc. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Trường hợp 1: - CPP theo quy định. - Báo cáo nghiên cứu tương đương sinh học theo hướng dẫn kỹ thuật của ASEAN ban hành theo Thông tư này và quy định tại Thông tư quy định thuốc phải thử tương đương sinh học và các yêu cầu đối với hồ sơ báo cáo số liệu nghiên cứu tương đương sinh học trong đăng ký lưu hành thuốc tại Việt Nam. 2. Trường hợp 2: - Bảng so sánh giữa thuốc được cấp giấy đăng ký lưu hành theo hình thức đăng ký lại và thuốc đã được công bố có tài liệu chứng minh tương đương sinh học (mẫu 12/TT). - Dữ liệu chứng minh các thay đổi không ảnh hưởng đến sinh khả dụng của thuốc trong trường hợp hồ sơ đăng ký lại có thay đổi ảnh hưởng không đáng kể đến sinh khả dụng của thuốc. 3. Trường hợp 3: - Bảng so sánh giữa thuốc được cấp giấy đăng ký lưu hành theo hình thức đăng ký lại và thuốc đã được công bố có tài liệu chứng minh tương đương sinh học (mẫu 12/TT). - Hồ sơ nghiên cứu tương đương sinh học theo quy định tại Thông tư này và quy định tại Thông tư quy định thuốc phải thử tương đương sinh học và các yêu cầu đối với hồ sơ báo cáo số liệu nghiên cứu tương đương sinh học trong đăng ký lưu hành thuốc tại Việt Nam. |
|
|
|
|
|
|
| Nội dung thay đổi 38 (MiV- PA38) | Cập nhật phân loại biệt dược gốc, sinh phẩm tham chiếu | x (TH1, 2, 3, 4, 5, 6, 7) | x (TH1, TH2 (2.3), TH3 (3.2), TH4 (4.3), TH5, TH6) |
|
| x (TH7, trừ trường hợp thuốc đã được cấp giấy đăng ký lưu hành theo bộ hồ sơ ACTD hoặc ICH-CTD bao gồm hồ sơ lâm sàng của thuốc) |
|
| Điều kiện cần đáp ứng (C) | Thuốc đề nghị phân loại biệt dược gốc, sinh phẩm tham chiếu khi đáp ứng điều kiện sau: 1. Trường hợp 1: Thuốc đã được Bộ Y tế công bố là biệt dược gốc, sinh phẩm tham chiếu được sản xuất toàn bộ tại nước có cơ quan quản lý thuộc danh sách quy định tại khoản 9 Điều 2 Thông tư này được phân loại biệt dược gốc, sinh phẩm tham chiếu nếu thuộc một trong các trường hợp sau đây: - Thuốc được cấp giấy đăng ký lưu hành mới theo hình thức đăng ký lại quy định tại Thông tư số 44/2014/TT- BYT mà có cùng công thức bào chế, quy trình sản xuất, tiêu chuẩn chất lượng nguyên liệu, tiêu chuẩn chất lượng thuốc thành phẩm so với biệt dược gốc đã được công bố hoặc có thay đổi liên quan đến nội dung trên đã được Cơ quan quản lý Việt Nam hoặc nước sở tại phê duyệt. - Thuốc thay đổi cơ sở sản xuất và được cấp giấy đăng ký lưu hành mới mà đáp ứng quy định tại điểm c khoản 1 Điều 9 Thông tư này. 2. Trường hợp 2: Thuốc sản xuất tại nước ngoài đã được Bộ Y tế công bố là biệt dược gốc, sinh phẩm tham chiếu không được sản xuất toàn bộ tại nước có cơ quan quản lý thuộc danh sách quy định tại khoản 9 Điều 2 Thông tư này nhưng được cấp phép lưu hành tại nước có cơ quan quản lý thuộc danh sách quy định tại khoản 9 Điều 2 Thông tư này được phân loại biệt dược gốc, sinh phẩm tham chiếu nếu thuộc một trong các trường hợp sau đây: - Thuốc có giấy đăng ký lưu hành còn hiệu lực hoặc được gia hạn hoặc được thay đổi, bổ sung không thuộc quy định tại điểm b khoản 2 Điều 55 Luật Dược; - Thuốc được cấp giấy đăng ký lưu hành mới theo hình thức đăng ký lại quy định tại Thông tư số 44/2014/TT- BYT mà có cùng công thức bào chế, quy trình sản xuất, tiêu chuẩn chất lượng nguyên liệu, tiêu chuẩn chất lượng thuốc thành phẩm so với biệt dược gốc đã được công bố hoặc có thay đổi liên quan đến nội dung trên đã được Cơ quan quản lý Việt Nam hoặc nước sở tại phê duyệt; - Thuốc thay đổi cơ sở sản xuất và được cấp giấy đăng ký lưu hành mới mà đáp ứng quy định tại điểm c khoản 1 Điều 9 Thông tư này. 3. Trường hợp 3: Thuốc đã được Bộ Y tế công bố là biệt dược gốc, sinh phẩm tham chiếu sản xuất toàn bộ các công đoạn tại Việt Nam hoặc sản xuất một, một số công đoạn tại Việt Nam và các công đoạn sản xuất còn lại được thực hiện toàn bộ tại nước có cơ quan quản lý thuộc danh sách quy định tại khoản 9 Điều 2 Thông tư này được phân loại biệt dược gốc, sinh phẩm tham chiếu nếu thuộc một trong các trường hợp sau đây: - Thuốc được cấp giấy đăng ký lưu hành mới theo hình thức đăng ký lại quy định tại Thông tư số 44/2014/TT- BYT mà có cùng công thức bào chế, quy trình sản xuất, tiêu chuẩn chất lượng nguyên liệu, tiêu chuẩn chất lượng thuốc thành phẩm so với biệt dược gốc đã được công bố hoặc có thay đổi liên quan đến nội dung trên đã được Cơ quan quản lý Việt Nam hoặc nước sở tại phê duyệt. - Thuốc thay đổi cơ sở sản xuất và được cấp giấy đăng ký lưu hành mới mà đáp ứng quy định tại điểm c khoản 1 Điều 9 Thông tư này. 4. Trường hợp 4: Thuốc đã được Bộ Y tế công bố là biệt dược gốc, sinh phẩm tham chiếu sản xuất một, một số công đoạn tại Việt Nam và các công đoạn sản xuất còn lại không được thực hiện toàn bộ tại nước có cơ quan quản lý thuộc danh sách quy định tại khoản 9 Điều 2 Thông tư này nhưng được cấp phép lưu hành tại nước có cơ quan quản lý thuộc danh sách quy định tại khoản 9 Điều 2 Thông tư này được tiếp tục phân loại biệt dược gốc, sinh phẩm tham chiếu nếu thuộc một trong các trường hợp sau đây: - Thuốc có giấy đăng ký lưu hành còn hiệu lực hoặc được gia hạn hoặc được thay đổi, bổ sung không thuộc trường hợp quy định tại điểm b khoản 2 Điều 55 Luật Dược; - Thuốc được cấp giấy đăng ký lưu hành mới theo hình thức đăng ký lại quy định tại Thông tư số 44/2014/TT- BYT mà có cùng công thức bào chế, quy trình sản xuất, tiêu chuẩn chất lượng nguyên liệu, tiêu chuẩn chất lượng thuốc thành phẩm so với biệt dược gốc đã được công bố hoặc có thay đổi liên quan đến nội dung trên đã được Cơ quan quản lý Việt Nam hoặc nước sở tại phê duyệt; - Thuốc thay đổi cơ sở sản xuất và được cấp giấy đăng ký lưu hành mới mà đáp ứng quy định tại điểm c khoản 1 Điều 9 Thông tư này. 5. Trường hợp 5: Thuốc đã được Bộ Y tế công bố là biệt dược gốc, sinh phẩm tham chiếu sản xuất toàn bộ tại nước có cơ quan quản lý thuộc danh sách quy định tại khoản 9 Điều 2 Thông tư này, được đặt gia công hoặc chuyển giao công nghệ sản xuất tại Việt Nam thì thuốc gia công hoặc chuyển giao công nghệ sản xuất một, một số hoặc toàn bộ các công đoạn sản xuất tại Việt Nam và cấp giấy đăng ký lưu hành mới được tiếp tục phân loại biệt dược gốc, sinh phẩm tham chiếu nếu đáp ứng quy định tại điểm b khoản 1 Điều 9 Thông tư này. 6. Trường hợp 6: Thuốc đã được Bộ Y tế công bố là biệt dược gốc, sinh phẩm tham chiếu không được sản xuất toàn bộ tại nước có cơ quan quản lý thuộc danh sách quy định tại khoản 9 Điều 2 Thông tư này nhưng được cấp phép lưu hành tại nước có cơ quan quản lý thuộc danh sách quy định tại khoản 9 Điều 2 Thông tư này, được gia công hoặc chuyển giao công nghệ sản xuất tại Việt Nam thì thuốc gia công hoặc chuyển giao công nghệ sản xuất một, một số hoặc toàn bộ các công đoạn sản xuất tại Việt Nam và cấp giấy đăng ký lưu hành mới được phân loại biệt dược gốc, sinh phẩm tham chiếu nếu đáp ứng quy định tại điểm b khoản 1 Điều 9 Thông tư này. 7. Trường hợp 7: Thuốc chưa được Bộ Y tế công bố biệt dược gốc, sinh phẩm tham chiếu nếu đáp ứng quy định tại điểm a khoản 1 Điều 9 Thông tư này thì được phân loại biệt dược gốc, sinh phẩm tham chiếu. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Trường hợp 1: Thuốc đã được Bộ Y tế công bố là biệt dược gốc, sinh phẩm tham chiếu được sản xuất toàn bộ tại nước có cơ quan quản lý thuộc danh sách quy định tại khoản 9 Điều 2 Thông tư này: 1.1 Thuốc được cấp giấy đăng ký lưu hành mới theo hình thức đăng ký lại quy định tại Thông tư số 44/2014/TT-BYT: - Cam kết theo Mẫu 11A/TT về thuốc đề nghị và thuốc biệt dược gốc đã được công bố. 1.2. Thuốc thay đổi cơ sở sản xuất và được cấp giấy đăng ký lưu hành mới: - Cam kết theo Mẫu 11C/TT về thuốc đề nghị và thuốc biệt dược gốc đã được công bố. - Tài liệu chứng minh theo Phụ lục II Thông tư này tương ứng với các nội dung thay đổi để chứng minh thuốc thay đổi cơ sở sản xuất tương đương về chất lượng so với biệt dược gốc trước khi thay đổi (nếu có thay đổi một trong các nội dung này). 2. Trường hợp 2: Thuốc sản xuất tại nước ngoài đã được Bộ Y tế công bố là biệt dược gốc, sinh phẩm tham chiếu không được sản xuất toàn bộ tại nước có cơ quan quản lý thuộc danh sách quy định tại khoản 9 Điều 2 Thông tư này 2.1. Thuốc có giấy đăng ký lưu hành còn hiệu lực hoặc được gia hạn giấy đăng ký lưu hành hoặc được thay đổi, bổ sung giấy đăng ký lưu hành không thuộc trường hợp quy định tại điểm b khoản 2 Điều 55 Luật Dược: - CPP được cấp phép lưu hành bởi một trong các cơ quan quản lý quy định tại khoản 9 Điều 2 Thông tư này. 2.2. Thuốc được cấp giấy đăng ký lưu hành mới theo hình thức đăng ký lại quy định tại Thông tư số 44/2014/TT-BYT: - CPP được cấp phép lưu hành bởi một các trong cơ quan quản lý quy định tại khoản 9 Điều 2 Thông tư này. - Cam kết theo Mẫu 11A/TT về thuốc đề nghị và thuốc biệt dược gốc đã được công bố 2.3. Thuốc thay đổi cơ sở sản xuất và được cấp giấy đăng ký lưu hành mới: - CPP được cấp phép lưu hành bởi một trong các cơ quan quản lý quy định tại khoản 9 Điều 2 Thông tư này. - Cam kết theo Mẫu 11C/TT về thuốc đề nghị và thuốc biệt dược gốc đã được công bố. - Tài liệu chứng minh theo Phụ lục II Thông tư này tương ứng với các nội dung thay đổi để chứng minh thuốc thay đổi cơ sở sản xuất tương đương về chất lượng so với biệt dược gốc trước khi thay đổi (nếu có thay đổi một trong các nội dung này). 3. Trường hợp 3. Thuốc đã được Bộ Y tế công bố là biệt dược gốc, sinh phẩm tham chiếu được sản xuất toàn bộ các công đoạn tại Việt Nam hoặc sản xuất một, một số công đoạn tại Việt Nam và các công đoạn sản xuất còn lại được thực hiện toàn bộ tại nước có cơ quan quản lý thuộc danh sách quy định tại khoản 9 Điều 2 Thông tư này: 3.1. Thuốc được cấp giấy đăng ký lưu hành mới theo hình thức đăng ký lại quy định tại Thông tư số 44/2014/TT-BYT: - Cam kết theo Mẫu 11A/TT về thuốc đề nghị và thuốc biệt dược gốc đã được công bố. 3.2. Thuốc thay đổi cơ sở sản xuất và được cấp giấy đăng ký lưu hành mới: - Cam kết theo Mẫu 11C/TT về thuốc đề nghị và thuốc biệt dược gốc đã được công bố. - Tài liệu chứng minh theo Phụ lục II Thông tư này tương ứng với các nội dung thay đổi để chứng minh thuốc thay đổi cơ sở sản xuất tương đương về chất lượng so với biệt dược gốc trước khi thay đổi (nếu có thay đổi một trong các nội dung này). 4. Trường hợp 4. Thuốc đã được Bộ Y tế công bố là biệt dược gốc, sinh phẩm tham chiếu được sản xuất một, một số công đoạn tại Việt Nam và các công đoạn sản xuất còn lại không được thực hiện toàn bộ tại nước có cơ quan quản lý thuộc danh sách quy định tại khoản 9 Điều 2 Thông tư này: 4.1. Thuốc có giấy đăng ký lưu hành còn hiệu lực hoặc được gia hạn giấy đăng ký lưu hành hoặc được thay đổi, bổ sung giấy đăng ký lưu hành không thuộc trường hợp quy định tại điểm b khoản 2 Điều 55 Luật Dược: - CPP được cấp phép lưu hành bởi một trong các cơ quan quản lý quy định tại khoản 9 Điều 2 Thông tư này. 4.2 Thuốc được cấp giấy đăng ký lưu hành mới theo hình thức đăng ký lại quy định tại Thông tư số 44/2014/TT-BYT: - CPP được cấp phép lưu hành bởi một trong các cơ quan quản lý quy định tại khoản 9 Điều 2 Thông tư này. - Cam kết theo Mẫu 11A/TT về thuốc đề nghị và thuốc biệt dược gốc đã được công bố. 4.3. Thuốc thay đổi cơ sở sản xuất và được cấp giấy đăng ký lưu hành mới: - CPP được cấp phép lưu hành bởi một trong các cơ quan quản lý quy định tại khoản 9 Điều 2 Thông tư này. - Cam kết theo Mẫu 11C/TT về thuốc đề nghị và thuốc biệt dược gốc đã được công bố. - Tài liệu chứng minh theo Phụ lục II Thông tư này tương ứng với các nội dung thay đổi để chứng minh thuốc thay đổi cơ sở sản xuất tương đương về chất lượng so với biệt dược gốc trước khi thay đổi (nếu có thay đổi một trong các nội dung này). 5. Trường hợp 5. Thuốc đã được Bộ Y tế công bố là biệt dược gốc, sinh phẩm tham chiếu được sản xuất toàn bộ tại nước có cơ quan quản lý thuộc danh sách quy định tại khoản 9 Điều 2 Thông tư này, được gia công hoặc chuyển giao công nghệ sản xuất tại Việt Nam và được cấp giấy đăng ký lưu hành mới: - Cam kết theo Mẫu 11B/TT về thuốc gia công hoặc chuyển giao công nghệ sản xuất tại Việt Nam và thuốc trước gia công hoặc chuyển giao công nghệ đã được công bố biệt dược gốc. - Tài liệu chứng minh theo Phụ lục II Thông tư này tương ứng với các nội dung thay đổi để chứng minh thuốc sản xuất tại Việt Nam tương đương về chất lượng so với biệt dược gốc trước khi gia công hoặc chuyển giao (nếu có thay đổi một trong các nội dung này). 6. Trường hợp 6. Thuốc đã được Bộ Y tế công bố là biệt dược gốc, sinh phẩm tham chiếu không được sản xuất toàn bộ tại nước có cơ quan quản lý thuộc danh sách quy định tại khoản 9 Điều 2 Thông tư này, gia công hoặc chuyển giao công nghệ sản xuất tại Việt Nam và được cấp giấy đăng ký lưu hành mới: - CPP thuốc trước gia công hoặc chuyển giao công nghệ được cấp phép lưu hành bởi một trong cơ quan quản lý quy định tại khoản 9 Điều 2 Thông tư này. - Cam kết theo Mẫu 11B/TT về thuốc gia công hoặc chuyển giao công nghệ sản xuất tại Việt Nam và thuốc trước chuyển giao công nghệ đã được công bố biệt dược gốc. - Tài liệu chứng minh theo Phụ lục II Thông tư này tương ứng với các nội dung thay đổi để chứng minh thuốc sản xuất tại Việt Nam tương đương về chất lượng so với biệt dược gốc trước khi gia công hoặc chuyển giao (nếu có thay đổi một trong các nội dung này). 7. Trường hợp 7. Thuốc chưa được Bộ Y tế công bố, đề nghị phân loại là biệt dược gốc, sinh phẩm tham chiếu: - CPP được cấp phép lưu hành bởi một trong các cơ quan quản lý quy định tại khoản 9 Điều 2 Thông tư này đối với thuốc không được sản xuất toàn bộ tại nước có cơ quan quản lý quy định tại khoản 9 Điều 2 Thông tư này (trừ thuốc sản xuất tại Việt Nam). - Tài liệu tiền lâm sàng và tài liệu lâm sàng (kèm theo tờ hướng dẫn sử dụng đã được phê duyệt), trừ trường hợp thuốc đã được cấp giấy đăng ký lưu hành theo bộ hồ sơ ACTD hoặc ICH-CTD bao gồm hồ sơ lâm sàng của thuốc. |
|
|
|
|
|
|
| Nội dung thay đổi 39 (MiV- PA39) | Bổ sung, cập nhật thông tin để cung cấp thông tin thuốc, quảng cáo thuốc |
|
|
|
|
|
|
| TH1 | Có hồ sơ lâm sàng | x |
|
| x | x |
|
| TH2 | Không có hồ sơ lâm sàng | x |
|
| x |
|
|
| Điều kiện cần đáp ứng (C) | 1. Thay đổi/bổ sung cần phê duyệt nhưng không dẫn đến thay đổi thông tin trên nhãn và hướng dẫn sử dụng thuốc (*) 2. Các thông tin được bổ sung, cập nhật theo quy định tại mục này là các thông tin mới được phát minh, phát hiện qua nghiên cứu khoa học hoặc qua theo dõi sản phẩm trên thị trường nhưng không thuộc trường hợp quy định tại mục từ 1 đến 7 Phụ lục này. 3. Trình bày toàn bộ các thông tin đề nghị theo đúng nội dung sẽ sử dụng để thông tin thuốc. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Nội dung thông tin đề nghị cập nhật. 2. Tài liệu tham khảo. 3. Bản sao tờ hướng dẫn sử dụng thuốc cập nhật nhất đã được Bộ Y tế phê duyệt có đóng dấu của cơ sở đăng ký thuốc. |
|
|
|
|
|
|
| Nội dung thay đổi 40 (MiV- PA40) | Thay đổi hoặc bổ sung cơ sở/địa điểm chịu trách nhiệm về kiểm nghiệm chất lượng | x | x |
|
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Chỉ áp dụng cho cơ sở, địa điểm kiểm nghiệm chất lượng. 2. Cơ sở sản xuất và cơ sở đóng gói thứ cấp của thuốc thành phẩm không thay đổi. 3. Việc chuyển giao phương pháp từ cơ sở/địa điểm đã được phê duyệt đến cơ sở/địa điểm mới hoặc phòng kiểm nghiệm mới đã được hoàn thành. |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Cam kết của cơ sở sản xuất sản phẩm/chủ sở hữu sản phẩm liên quan các nội dung sau: a) Việc thay đổi không ảnh hưởng đến tiêu chuẩn chất lượng thuốc thành phẩm. b) Các phép thử do cơ sở/địa điểm mới hoặc phòng kiểm nghiệm mới sử dụng tương đương với các phương pháp đã được phê duyệt. c) Danh sách các phép thử do cơ sở/địa điểm mới hoặc phòng kiểm nghiệm mới sử dụng kèm theo xác nhận về việc đã hoàn tất việc chuyển giao phương pháp cho mỗi phép thử. 2. Tài liệu pháp lý chứng minh cơ sở/địa điểm mới hoặc phòng kiểm nghiệm mới đã được công nhận phù hợp (GMP/GLP/ISO/IEC). 3. Thư ủy quyền của chủ sở hữu sản phẩm cho cơ sở/địa điểm mới hoặc phòng kiểm nghiệm chịu trách nhiệm về kiểm nghiệm chất lượng thuốc thành phẩm mới (nếu có). 4. Dữ liệu chuyển giao phương pháp phân tích/dữ liệu xác minh (nếu có). 5. Phần hồ sơ S2 hoặc P3 (ACTD) cập nhật các nội dung thay đổi |
|
|
|
|
|
|
| Nội dung thay đổi 41 (MiV-PA41) | Danh mục thuốc sản xuất trong nước được cấp phép lưu hành bởi một trong các cơ quan quản lý dược của nước thuộc danh sách SRA | x | x | x |
|
|
|
| Điều kiện cần đáp ứng (C) | 1. Thuốc sản xuất toàn bộ trên dây chuyền sản xuất tại Việt Nam. 2. Thuốc được cấp phép lưu hành bởi cơ quan quản lý SRA quy định tại khoản 9 Điều 2 Thông tư này có cùng các tiêu chí sau: công thức bào chế, quy trình sản xuất, tiêu chuẩn chất lượng, phương pháp kiểm nghiệm; dược chất, tá dược phải có cùng tiêu chuẩn chất lượng, cơ sở sản xuất, địa điểm sản xuất |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | 1. Giấy tờ pháp lý do cơ quan quản lý dược của nước thuộc danh sách SRA cấp gồm nội dung tối thiểu sau đây: tên thuốc, hoạt chất, nồng độ hoặc hàm lượng hoạt chất, dạng bào chế, tên và địa chỉ cơ sở sản xuất, có xác nhận thuốc được cấp phép lưu hành tại nước đó. 2. Bảng kê khai các thông tin để chứng minh thuốc lưu hành tại Việt Nam và thuốc được nước thuộc danh sách SRA cấp phép lưu hành có cùng công thức bào chế, quy trình sản xuất, tiêu chuẩn chất lượng, phương pháp kiểm nghiệm; dược chất, tá dược phải có cùng tiêu chuẩn chất lượng, cơ sở sản xuất, địa điểm sản xuất. |
|
|
|
|
|
|
| Nội dung thay đổi 42 (MiV- PA42) | Danh mục thuốc có đăng ký sử dụng nguồn nguyên liệu (dược chất) được cấp giấy chứng nhận CEP để sản xuất | x | x |
|
|
|
|
| Điều kiện cần đáp ứng (C) | Thuốc có đăng ký sử dụng nguồn nguyên liệu (dược chất) được cấp giấy chứng nhận CEP để sản xuất |
|
|
|
|
|
|
| Hồ sơ cần nộp (D) | Giấy chứng nhận CEP (Certificate of Suitability to the Monographs of the European Pharmacopoeia) của nguyên liệu. |
|
|
|
|
|
|
C. VẮC XIN
|
| Nội dung thay đổi | Pháp chế | Tiêu chuẩn | Bào chế | Dược lý | Lâm sàng |
| Nội dung thay đổi 1 (MiV- PA1.C) | Cập nhật chủng cúm mùa hàng năm | x | x | x | x | x |
| Điều kiện đáp ứng (C) | Không có |
|
|
|
|
|
| Hồ sơ cần nộp (D) | Phần I (Hành chính): - Đơn đăng ký (theo mẫu) - Giấy chứng nhận sản phẩm dược phẩm (CPP). - Giấy chứng nhận xuất xưởng lô hoặc giấy chứng nhận kết quả kiểm định được cấp bởi cơ quan có thẩm quyền nước cấp CPP (là nước sản xuất có cơ quan quản lý đã được WHO công nhận đạt hệ thống quản lý quốc gia theo tiêu chuẩn của WHO về vắc xin (National Regulatory Authority - NRA) hoặc nước có cơ quan quản lý theo quy định tại khoản 9 Điều 2 Thông tư này). Trường hợp không cung cấp được, yêu cầu cung cấp giấy chứng nhận kết quả kiểm định chất lượng, độ an toàn chung của Viện Kiểm định quốc gia vắc xin và sinh phẩm y tế (NICVB). - Tài liệu về xuất xứ chủng gốc. - Khuyến cáo của Tổ chức Y tế thế giới. - Mẫu nhãn, tờ hướng dẫn sử dụng đã được Cục Quản lý Dược phê duyệt. - Mẫu nhãn, tờ hướng dẫn sử dụng cập nhật thông tin chủng cúm mới. Phần II: Hồ sơ chất lượng liên quan theo quy định tại mục C.2 Phụ lục này. |
|
|
|
|
|
C. THUỐC DƯỢC LIỆU, NGUYÊN LIỆU LÀM THUỐC DƯỢC LIỆU
Thực hiện theo quy định tại Mục A Phụ lục này (kèm theo giải trình phù hợp trong trường hợp không cung cấp được đầy đủ các tài liệu cần nộp theo quy định do đặc thù riêng của thuốc dược liệu) hoặc các thay đổi quy định riêng cho thuốc dược liệu như sau:
1. THAY ĐỔI LỚN
|
| Nội dung thay đổi | Pháp chế | Tiêu chuẩn | Bào chế |
| Nội dung thay đổi 1 (MaV-1.D) | Thay đổi/bổ sung nguồn dược liệu | x | x | x |
| Điều kiện đáp ứng (C) | Thay đổi, bổ sung theo hướng nguồn dược liệu mới được kiểm soát chất lượng tương đương hoặc ở mức cao hơn |
|
|
|
| Hồ sơ cần nộp (D) | Phần I (Hành chính) - Đơn đăng ký (theo mẫu) - Giấy tờ pháp lý của cơ sở sản xuất bán thành phẩm dược liệu và dược liệu mới theo quy định tại khoản 11 Điều 22 Thông tư này. Phần II (Chất lượng): - Tài liệu chất lượng theo quy định tại khoản 1 Điều 28 Thông tư này; - Phiếu kiểm nghiệm của thuốc thành phẩm; - Nghiên cứu độ ổn định của thuốc thành phẩm theo quy định như đối với thuốc hóa dược thay đổi/bổ sung nguồn nguyên liệu. |
|
|
|
2. THAY ĐỔI NHỎ PHẢI ĐƯỢC PHÊ DUYỆT CỦA CƠ QUAN QUẢN LÝ
| STT | Nội dung thay đổi/bổ sung | Pháp chế | Tiêu chuẩn | Bào chế | Dược lý | Lâm sàng |
| Nội dung thay đổi 1 (MiV- PA1.D) | Thay đổi chất chuẩn để kiểm nghiệm nguyên liệu | x | x |
|
|
|
| Điều kiện đáp ứng (C) | Tiêu chuẩn chất lượng nguyên liệu không đổi ngoại trừ chất chuẩn sử dụng. |
|
|
|
|
|
| Hồ sơ cần nộp (D) | Phần I (Hành chính): - Đơn đăng ký (theo mẫu) Phần II (Chất lượng): - Giải trình lý do, căn cứ khoa học đối với nội dung thay đổi - Bảng so sánh tiêu chuẩn chất lượng thành phẩm đã được phê duyệt và sau khi thay đổi chất chuẩn - Bản tiêu chuẩn chất lượng thành phẩm thay đổi - Phiếu kiểm nghiệm thành phẩm được kiểm tra theo tiêu chuẩn chất lượng thành phẩm sau khi đã thay đổi chất chuẩn - Phiếu kiểm nghiệm của chất chuẩn mới xin thay đổi - Các tài liệu liên quan khác (nếu có) |
|
|
|
|
|
| Nội dung thay đổi 2 (MiV- PA2.D) | Thay đổi/bổ sung/bỏ bớt nhà sản xuất bao bì | x | x |
|
|
|
| Điều kiện đáp ứng (C) | - Không làm thay đổi chất lượng và độ ổn định của thuốc |
|
|
|
|
|
| Hồ sơ cần nộp (D) | Phần I (Hành chính): - Đơn đăng ký (theo mẫu) Phần II (Chất lượng): - Tiêu chuẩn chất lượng bao bì (nếu có thay đổi) - Phiếu kiểm nghiệm bao bì |
|
|
|
|
|
| Nội dung thay đổi 3 (MiV- PA3.D) | Thay đổi tiêu chuẩn chất lượng bán thành phẩm dược liệu | x | x |
|
|
|
| Điều kiện đáp ứng (C) | - Chỉ áp dụng cho trường hợp tiêu chuẩn chất lượng đã được duyệt của bán thành phẩm dược liệu không theo một trong các dược điển tham chiếu. - Thay đổi nhằm đáp ứng theo những thay đổi trong tiêu chuẩn chất lượng dược liệu tương ứng trong công thức bào chế bán thành phẩm dược liệu khi cập nhật/thay đổi theo phiên bản mới của dược điển (trường hợp tiêu chuẩn chất lượng dược liệu theo tiêu chuẩn dược điển) hoặc cập nhật các chỉ tiêu chất lượng theo dược điển tham khảo (trường hợp tiêu chuẩn chất lượng dược liệu không theo tiêu chuẩn dược điển). |
|
|
|
|
|
| Hồ sơ cần nộp (D) | Phần I (Hành chính): - Đơn đăng ký (theo mẫu) Phần II (Chất lượng): - Bảng so sánh tiêu chuẩn chất lượng dược liệu đã được phê duyệt và tiêu chuẩn đề nghị thay đổi/cập nhật. - Phiếu kiểm nghiệm dược liệu theo tiêu chuẩn chất lượng cập nhật/thay đổi. - Bảng so sánh tiêu chuẩn chất lượng bán thành phẩm dược liệu đã được duyệt và tiêu chuẩn đề nghị thay đổi. - Bảng so sánh phân tích lô bán thành phẩm dược liệu theo tiêu chuẩn chất lượng đã được phê duyệt và tiêu chuẩn thay đổi. - Bản tiêu chuẩn chất lượng đề nghị thay đổi của dược liệu và bán thành phẩm dược liệu. - Đối với thay đổi quy trình phân tích trong tiêu chuẩn chất lượng phù hợp với các thay đổi theo tiêu chất lượng mới cập nhật/thay đổi của dược liệu, yêu cầu dữ liệu thẩm tra quy trình phân tích mới. |
|
|
|
|
|
* Ghi chú: Trên là nguyên tắc chung để chuyển hồ sơ thẩm định. Bên cạnh đó, Chuyên viên phụ trách cần căn cứ tài liệu nộp trong hồ sơ để chuyển các tiểu ban thẩm định cho phù hợp.
BM.ĐK.08.08/13
Danh mục hồ sơ bản giấy cần chuyển CG, đơn vị thẩm định
DANH MỤC HỒ SƠ BẢN GIẤY CHUYỂN CHUYÊN GIA, ĐƠN VỊ THẨM ĐỊNH
| Stt | Ngày nộp | Mã HS | Tên thuốc | Công ty ĐK | CG/Đơn vị thẩm định yêu cầu | Lý do yêu cầu | Hồ sơ lần đầu/Hồ sơ bổ sung | Ghi chú |
| 1 |
|
|
|
|
|
|
|
|
| 2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Tổng số:
Ngày…………tháng………năm………..
| Chuyên viên phụ trách | Ý kiến của Lãnh đạo Phòng |
- 1Quyết định 453/QĐ-QLD năm 2023 về Quy trình thao tác chuẩn trong Hệ thống quản lý chất lượng theo tiêu chuẩn ISO 9001:2015 áp dụng vào hoạt động quản lý nhà nước do Cục trưởng Cục Quản lý Dược ban hành
- 2Quyết định 693/QĐ-QLD năm 2023 về Quy trình thao tác chuẩn trong Hệ thống quản lý chất lượng theo tiêu chuẩn ISO 9001:2015 áp dụng vào hoạt động quản lý nhà nước tại Cục Quản lý Dược
- 3Quyết định 742/QĐ-QLD năm 2023 về Quy trình thao tác chuẩn trong Hệ thống quản lý chất lượng theo tiêu chuẩn ISO 9001:2015 áp dụng vào hoạt động quản lý nhà nước tại Cục Quản lý Dược
- 4Quyết định 804/QĐ-QLD năm 2023 về Quy trình thao tác chuẩn trong Hệ thống quản lý chất lượng theo tiêu chuẩn ISO 9001:2015 áp dụng vào hoạt động quản lý nhà nước tại Cục Quản lý Dược
- 5Quyết định 178/QĐ-QLD năm 2024 về Quy trình thao tác chuẩn trong Hệ thống quản lý chất lượng theo tiêu chuẩn ISO 9001:2015 áp dụng vào hoạt động quản lý Nhà nước tại Cục Quản lý Dược
- 1Thông tư 05/2010/TT-BYT hướng dẫn bảo mật dữ liệu thử nghiệm trong đăng ký thuốc do Bộ Y tế ban hành
- 2Thông tư 44/2014/TT-BYT quy định việc đăng ký thuốc do Bộ trưởng Bộ Y tế ban hành
- 3Luật Dược 2016
- 4Thông tư 01/2018/TT-BYT về quy định ghi nhãn thuốc, nguyên liệu làm thuốc và tờ hướng dẫn sử dụng thuốc do Bộ trưởng Bộ Y tế ban hành
- 5Nghị định 54/2017/NĐ-CP hướng dẫn Luật dược do Chính phủ ban hành
- 6Nghị định 75/2017/NĐ-CP quy định chức năng, nhiệm vụ, quyền hạn và cơ cấu tổ chức của Bộ Y tế
- 7Nghị định 155/2018/NĐ-CP sửa đổi quy định liên quan đến điều kiện đầu tư kinh doanh thuộc phạm vi quản lý nhà nước của Bộ Y tế
- 8Thông tư 32/2018/TT-BYT quy định việc đăng ký lưu hành thuốc, nguyên liệu thuốc do Bộ trưởng Bộ Y tế ban hành
- 9Thông tư 08/2022/TT-BYT quy định việc đăng ký lưu hành thuốc, nguyên liệu làm thuốc do Bộ trưởng Bộ Y tế ban hành
- 10Nghị định 95/2022/NĐ-CP quy định chức năng, nhiệm vụ, quyền hạn và cơ cấu tổ chức của Bộ Y tế
- 11Quyết định 1969/QĐ-BYT năm 2023 quy định chức năng, nhiệm vụ, quyền hạn và cơ cấu tổ chức của Cục Quản lý Dược thuộc Bộ Y tế
- 12Quyết định 453/QĐ-QLD năm 2023 về Quy trình thao tác chuẩn trong Hệ thống quản lý chất lượng theo tiêu chuẩn ISO 9001:2015 áp dụng vào hoạt động quản lý nhà nước do Cục trưởng Cục Quản lý Dược ban hành
- 13Thông tư 16/2023/TT-BYT quy định về đăng ký lưu hành đối với thuốc gia công, thuốc chuyển giao công nghệ tại Việt Nam do Bộ trưởng Bộ Y tế ban hành
- 14Quyết định 693/QĐ-QLD năm 2023 về Quy trình thao tác chuẩn trong Hệ thống quản lý chất lượng theo tiêu chuẩn ISO 9001:2015 áp dụng vào hoạt động quản lý nhà nước tại Cục Quản lý Dược
- 15Quyết định 742/QĐ-QLD năm 2023 về Quy trình thao tác chuẩn trong Hệ thống quản lý chất lượng theo tiêu chuẩn ISO 9001:2015 áp dụng vào hoạt động quản lý nhà nước tại Cục Quản lý Dược
- 16Quyết định 804/QĐ-QLD năm 2023 về Quy trình thao tác chuẩn trong Hệ thống quản lý chất lượng theo tiêu chuẩn ISO 9001:2015 áp dụng vào hoạt động quản lý nhà nước tại Cục Quản lý Dược
- 17Quyết định 178/QĐ-QLD năm 2024 về Quy trình thao tác chuẩn trong Hệ thống quản lý chất lượng theo tiêu chuẩn ISO 9001:2015 áp dụng vào hoạt động quản lý Nhà nước tại Cục Quản lý Dược
Quyết định 658/QĐ-QLD năm 2023 về Quy trình thao tác chuẩn trong Hệ thống quản lý chất lượng theo tiêu chuẩn ISO 9001:2015 áp dụng vào hoạt động quản lý nhà nước tại Cục Quản lý Dược
- Số hiệu: 658/QĐ-QLD
- Loại văn bản: Quyết định
- Ngày ban hành: 19/09/2023
- Nơi ban hành: Cục Quản lý dược
- Người ký: Vũ Tuấn Cường
- Ngày công báo: Đang cập nhật
- Số công báo: Đang cập nhật
- Ngày hiệu lực: 19/09/2023
- Tình trạng hiệu lực: Kiểm tra
Đơn vị chủ quản: Công ty cổ phần tư vấn đầu tư và ứng dụng công nghệ 4.0.
Chịu trách nhiệm chính: Bà Phạm Hoài Thương.
Giấy chứng nhận ĐKDN số: 0108234370, do Sở Kế hoạch và Đầu tư thành phố Hà Nội cấp ngày 18/04/2018.
Địa chỉ: Thôn Trung, Xã Phù Đổng, TP Hà Nội - VPGD: C2 Vincom, 119 Trần Duy Hưng, Phường Yên Hòa, TP Hà Nội.
Điện thoại: 024.6294.9155 - Hotline: 0984.988.691 - Email: info@hethongphapluat.com