
Để sử dụng toàn bộ tiện ích nâng cao của Hệ Thống Pháp Luật vui lòng lựa chọn và đăng ký gói cước.
| BỘ Y TẾ | CỘNG HÒA XÃ HỘI CHỦ NGHĨA VIỆT NAM |
| Số: 4021/2003/QĐ-BYT | Hà Nội, ngày 30 tháng 07 năm 2003 |
VỀ VIỆC BAN HÀNH “QUY ĐỊNH ĐÁNH GIÁ VỆ SINH AN TOÀN 20 CHẤT PHỤ GIA THỰC PHẨM”
BỘ TRƯỞNG BỘ Y TẾ
Căn cứ Nghị định số 49/CP ngày 15/5/2003 của Chính phủ quy định chức năng nhiệm vụ, quyền hạn và cơ cấu tổ chức của Bộ Y tế;
Căn cứ Nghị định số 86/CP ngày 08/12/1995 của Chính phủ về việc phân công trách nhiệm quản lý nhà nước đối với chất lượng hàng hóa;
Căn cứ Quyết định số 14/1999/QĐ-TTg ngày 14/02/1999 của Thủ tướng Chính phủ về việc thành lập Cục Quản lý chất lượng vệ sinh an toàn thực phẩm thuộc Bộ Y tế;
Theo đề nghị của Vụ trưởng Vụ Khoa học đào tạo, Vụ trưởng Vụ Pháp chế - Bộ Y tế và Cục trưởng Cục An toàn vệ sinh thực phẩm,
QUYẾT ĐỊNH
Điều 1. Ban hành kèm theo Quyết định này “Quy định đánh giá vệ sinh an toàn 20 chất phụ gia thực phẩm”.
Điều 2. Quyết định này có hiệu lực sau 15 ngày, kể từ ngày đăng Công báo.
Điều 3. Cục trưởng Cục An toàn vệ sinh thực phẩm có trách nhiệm tổ chức, chỉ đạo, hướng dẫn triển khai và kiểm tra việc thực hiện Quyết định này.
Điều 4. Các Ông, Bà: Chánh Văn phòng; Chánh Thanh tra, Vụ trưởng Khoa học đào tạo, Vụ trưởng Vụ Pháp chế - Bộ Y tế, Cục trưởng Cục An toàn vệ sinh thực phẩm; Giám đốc Sở Y tế tỉnh, thành phố trực thuộc Trung ương, Thủ trưởng các đơn vị trực thuộc Bộ Y tế, Thủ trưởng Y tế ngành chịu trách nhiệm thi hành Quyết định này.
|
| KT. BỘ TRƯỞNG BỘ Y TẾ |
ĐÁNH GIÁ VỆ SINH AN TOÀN 20 CHẤT PHỤ GIA THỰC PHẨM
(ban hành kèm theo Quyết định số 4021/2003/QĐ-BYT ngày 30 tháng 7 năm 2003 của Bộ trưởng Bộ Y tế)
1. Phạm vi điều chỉnh: Quy định này đưa ra các yêu cầu bắt buộc về vệ sinh an toàn đối với các chất phụ gia thực phẩm, được sản xuất, nhập khẩu và lưu thông trên lãnh thổ Việt Nam.
2. Đối tượng áp dụng: Quy định này bắt buộc áp dụng đối với các tổ chức cá nhân sản xuất, nhập khẩu, kinh doanh phụ gia thực phẩm trên lãnh thổ Việt Nam.
3. Giải thích từ ngữ: Trong Quy định này các từ ngữ dưới đây được hiểu như sau:
a. Phụ gia thực phẩm: Là những chất không được coi là thực phẩm hoặc một thành phần của thực phẩm. Phụ gia thực phẩm có ít hoặc không có giá trị dinh dưỡng, được chủ động cho vào với mục đích đáp ứng yêu cầu công nghệ trong quá trình sản xuất, chế biến, xử lý, bao gói, vận chuyển và bảo quản thực phẩm. Phụ gia thực phẩm không bao gồm các chất ô nhiễm, các chất bổ sung vào thực phẩm với mục đích tăng thêm giá trị dinh dưỡng của thực phẩm.
Phụ gia thực phẩm trong danh mục là các chất phụ gia trong Quy định danh mục các chất phụ gia được phép sử dụng trong thực phẩm theo quy định hiện hành của Bộ Y tế;
b. Yêu cầu vệ sinh an toàn đối với phụ gia thực phẩm: Là việc đảm bảo các phụ gia thực phẩm được sản xuất, nhập khẩu, kinh doanh có hàm lượng các chất độc hại, định lượng chất chính không vượt quá giới hạn cho phép theo Quy định này;
c. Phương pháp thử: Là các phương pháp phát hiện, xác định hàm lượng chất chính, đánh giá hàm lượng các chất độc hại trong sản phẩm phụ gia thực phẩm đáp ứng yêu cầu vệ sinh an toàn theo Quy định này.
4. Các tổ chức cá nhân chỉ được phép sản xuất, nhập khẩu, kinh doanh các phụ gia thực phẩm trong danh mục theo quy định hiện hành của Bộ Y tế và có chứng nhận phù hợp các yêu cầu về vệ sinh an toàn theo Quy định này do cơ quan có thẩm quyền cấp.
1. Quy định đánh giá vệ sinh an toàn Acid sorbic Phụ lục 1
2. Quy định đánh giá vệ sinh an toàn Kali sorbat Phụ lục 2
3. Quy định đánh giá vệ sinh an toàn Calci sorbat Phụ lục 3
4. Quy định đánh giá vệ sinh an toàn Natri benzoat Phụ lục 4
5. Quy định đánh giá vệ sinh an toàn Kali benzoat Phụ lục 5
6. Quy định đánh giá vệ sinh an toàn Etyl p-hydroxybenzoat Phụ lục 6
7. Quy định đánh giá vệ sinh an toàn Propyl p-hydroxybenzoat Phụ lục 7
8. Quy định đánh giá vệ sinh an toàn Metyl p-hydroxybenzoat Phụ lục 8
9. Quy định đánh giá vệ sinh an toàn Kali nitrat Phụ lục 9
10. Quy định đánh giá vệ sinh an toàn ter-Butyl hydroquinon (TBHQ) Phụ lục 10
11. Quy định đánh giá vệ sinh an toàn Butyl hydroxyanisol (BHA) Phụ lục 11
12. Quy định đánh giá vệ sinh an toàn Butyl hydroxytoluen (BHT) Phụ lục 12
13. Quy định đánh giá vệ sinh an toàn Mannitol Phụ lục 13
14. Quy định đánh giá vệ sinh an toàn Aspartam Phụ lục 14
15. Quy định đánh giá vệ sinh an toàn Isomalt Phụ lục 15
16. Quy định đánh giá vệ sinh an toàn Sucraloza Phụ lục 16
17. Quy định đánh giá vệ sinh an toàn Red 2G Phụ lục 17
18. Quy định đánh giá vệ sinh an toàn Green S Phụ lục 18
19. Quy định đánh giá vệ sinh an toàn Brilliant black Phụ lục 19
20. Quy định đánh giá vệ sinh an toàn Brown HT Phụ lục 20
III. HƯỚNG DẪN CHUNG CÁC PHƯƠNG PHÁP THỬ
Các phương pháp thử được áp dụng cho việc đánh giá hàm lượng các chất độc hại của 20 chất phụ gia thực phẩm tại mục 2 nêu trên (Phụ lục 21).
Các tổ chức cá nhân sản xuất, nhập khẩu, kinh doanh phụ gia thực phẩm vi phạm Quy định này, tuỳ theo mức độ vi phạm sẽ bị xử lý vi phạm hành chính hoặc bị truy cứu trách nhiệm hình sự, nếu gây thiệt hại thì phải bồi thường theo quy định của pháp luật.
|
| KT. BỘ TRƯỞNG BỘ Y TẾ |
QUY ĐỊNH ĐÁNH GIÁ VỆ SINH AN TOÀN CÁC CHẤT PHỤ GIA THỰC PHẨM
Phụ lục 1. Quy định đánh giá vệ sinh an toàn Acid sorbic
Phụ lục 2. Quy định đánh giá vệ sinh an toàn Kali sorbat
Phụ lục 3. Quy định đánh giá vệ sinh an toàn Calci sorbat
Phụ lục 4. Quy định đánh giá vệ sinh an toàn Natri benzoat
Phụ lục 5. Quy định đánh giá vệ sinh an toàn Kali benzoat
Phụ lục 6. Quy định đánh giá vệ sinh an toàn Etyl p-hydroxybenzoat
Phụ lục 7. Quy định đánh giá vệ sinh an toàn Propyl p-hydroxybenzoat
Phụ lục 8. Quy định đánh giá vệ sinh an toàn Metyl p-hydroxybenzoat
Phụ lục 9. Quy định đánh giá vệ sinh an toàn Kali nitra
Phụ lục 10. Quy định đánh giá vệ sinh an toàn ter-Butyl hydroquinon (TBHQ)
Phụ lục 11. Quy định đánh giá vệ sinh an toàn Butyl hydroxyanisol (BHA)
Phụ lục 12. Quy định đánh giá vệ sinh an toàn Butyl hydroxytoluen (BHT)
Phụ lục 13. Quy định đánh giá vệ sinh an toàn Mannitol
Phụ lục 14. Quy định đánh giá vệ sinh an toàn Aspartam
Phụ lục 15. Quy định đánh giá vệ sinh an toàn Isomalt
Phụ lục 16. Quy định đánh giá vệ sinh an toàn Sucraloza
Phụ lục 17. Quy định đánh giá vệ sinh an toàn Red 2G
Phụ lục 18. Quy định đánh giá vệ sinh an toàn Green S
Phụ lục 19. Quy định đánh giá vệ sinh an toàn Brilliant black
Phụ lục 20. Quy định đánh giá vệ sinh an toàn Brown HT
Phụ lục 21. Hướng dẫn chung các phương pháp thử
P.VL. Hướng dẫn phương pháp thử các tính chất vật lý
P.VL.01 Xác định khoảng nhiệt độ nóng chảy
P.VL.02 Xác định nhiệt độ đông đặc
P.VL.03 Xác định góc quay cực riêng
P.VC. Hướng dẫn phương pháp xác định các thành phần vô cơ
P.VC.01 Xác định giảm khối lượng khi làm khô
P.VC.02 Xác định giảm khối lượng khi nung
P.VC.03 Xác định hàm lượng nước (phương pháp Karl-fischer)
P.VC.04 Xác định hàm lượng clorid
P.VC.05 Xác định hàm lượng sulfat
P.VC.06 Xác định hàm lượng flo
P.VC.07 Xác định hàm lượng chì
P.VC.08 Xác định hàm lượng asen
P.VC.09 Xác định hàm lượng một số kim loại nặng bằng phương pháp Quang phổ hấp thụ nguyên tử (AAS)
P.VC.10 Xác định hàm lượng tổng các kim loại nặng
P.VC.11 Xác định tro sulfat
P.HC. Hướng dẫn các phương pháp xác định thành phần hữu cơ
P.HC.01 Xác định các hợp chất hữu cơ clo
P.HC.02 Xác định các hợp chất hữu cơ dễ cacbon hóa
P.HC.03 Xác định các hợp chất khử
P.PM. Hướng dẫn phương pháp xác định cho phẩm màu thực phẩm
P.PM.01 Định tính các chất màu
P.PM.02 Xác định giảm khối lượng khi sấy
P.PM.03 Xác định hàm lượng clorid
P.PM.04 Xác định hàm lượng sulfat
P.PM.05 Xác định hàm lượng các chất không tan trong nước
P.PM.06 Xác định hàm lượng các chất màu phụ
P.PM.07 Xác định hàm lượng các chất hữu cơ ngoài chất tạo màu
P.PM.08 Xác định hàm lượng các chất có thể chiết bằng ete.
P.PM.09 Xác định hàm lượng các amin thơm bậc nhất không sulphonat hóa
P.PM.10 Xác định hàm lượng leuso base trong chất màu triarylmetan sulphonat hóa
P.PM.11 Định lượng thành phần màu chính
TT. Các dung dịch thuốc thử
QUY ĐỊNH ĐÁNH GIÁ VỆ SINH AN TOÀN ACID SORBIC
1. Tên khác, ký hiệu Chỉ số INS: 200; Chỉ số EEC: E200
2. Định danh
Tên hoá học Acid sorbic; Acid 2,4 hexadienoic; acid 2-propenylacrylic
Mã số C.A.S 110-44-1
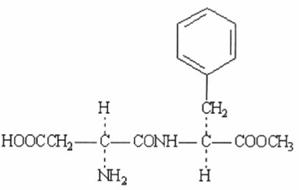
Công thức hoá học C6H8O2
| Công thức phân tử |
|
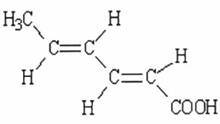
Khối lượng phân tử 111,12
3. Mô tả ngoại quan Tinh thể hình kim không màu hoặc bột mịn màu trắng, có mùi đặc trưng nhẹ.
4. Chức năng Bảo quản chống vi sinh vật, chống nấm mốc.
5. Yêu cầu về vệ sinh an toàn
5.1. Định tính
Tính tan Tan ít trong nước, tan trong etanol
Khoảng nhiệt độ nóng chảy 1320C - 1350C
Quang phổ Dung dịch mẫu thử trong isopropnol (1 trong 400.000) phải có độ hấp thụ quang cực đại tại bước sóng 254 ± 2nm
Định tính liên kết đôi Đạt (mô tả trong phần phương pháp thử)
5.2. Độ tinh khiết
Hàm lượng nước Không lớn hơn 0,5%
Trosulfat Không lớn hơn 0,2%
Asen Không lớn hơn 3mg/kg
Tổng các kim loại nặng Không lớn hơn 10mg/kg
Aldehyd Không lớn hơn 0,1%
5.3. Định lượng C6H8O2 Không nhỏ hơn 99,0% C6H8O2 (tính theo chế phẩm khan)
6. Phương pháp thử
6.1. Thử định tính
Xác định khoảng nhiệt độ nóng chảy
Xem hướng dẫn chung các phương pháp thử (P.VL.01)
Chú ý: Thiết bị đo độ chảy cần được gia nhiệt trước tới 1250C.
Đo quang phổ
Dung dịch mẫu thử trong isopropnol (1 trong 400.000) có độ hấp thụ quang cực đại tại bước sóng 254 ± 2nm
Thử định tính liên kết đôi
Lắc khoảng 0,02g mẫu với 1ml dung dịch brom (nước brom) TT.048, màu nâu của nước brom sẽ mất.
6.2. Thử độ tinh khiết
Xác định hàm lượng nước
Phương pháp chuẩn độ Karl-Fischer
Xem hướng dẫn chung các phương pháp thử (P.VC.03).
Xác định tro sulfat
Xem hướng dẫn chung các phương pháp thử (P.VC.11).
Chú ý: lấy 2g mẫu thử
Xác định Asen
Xem hướng dẫn chung các phương pháp thử (P.VC.08).
Xác định tổng các kim loại nặng
Xem hướng dẫn chung các phương pháp thử (P.VC.10 - Phương pháp 2).
Chú ý: Lấy 2g mẫu thử
Xác định aldehyd
Tiến hành:
- Ống thử: Lấy 1ml dung dịch mẫu bão hòa trong nước cất cho vào ống nghiệm và thêm vào đó 0,5ml dung dịch thuốc thử Schif TT.211, để yên trong 10 đến 15 phút.
- Ống chuẩn: Lấy 1ml dung dịch formaldehyd có nồng độ 2μg/ml cho vào ống nghiệm, thêm vào đó 0,5ml dung dịch thuốc thử Schiff TT.211, để yên trong 10 đến 15 phút.
- Màu của ống thử không được đậm hơn màu của ống chuẩn.
6.3. Định lượng C6H8O2
Phương pháp chuẩn độ trung hòa
Hòa tan 0,25g mẫu (chính xác đến 0,1mg) vào trong 50ml metanol khan, dung dịch metanol đã được trung hòa trước bằng dung dịch NaOH 0,1N với chỉ thị là dung dịch phenolphtalein TT.179. Chuẩn độ dung dịch mẫu bằng NaOH 0,1N cho đến khi dung dịch có màu hồng bền trong 30 giây. 1ml dung dịch NaOH 0,1N tương đương với 11,21mg C6H8O2
QUY ĐỊNH ĐÁNH GIÁ VỆ SINH AN TOÀN KALI SORBAT
1. Tên khác, ký hiệu Chỉ số INS: 202; Chỉ số EEC: E202
2. Định danh
Kali sorbat, kali (E.E) - 2,4 - hexadienoat; muối kali của acid trans 2,4-hexadienoic
Mã số C.A.S 24634-61-5
Công thức hoá học C6H7KO2
| Công thức phân tử |
|
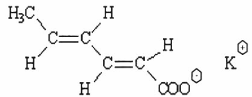
Khối lượng phân tử 150,22
3. Mô tả ngoại quan Tinh thể trắng hoặc trắng vàng, hoặc dạng bột tinh thể.
4. Chức năng Chất bảo quản
5. Yêu cầu về vệ sinh an toàn
5.1. Định tính
Tính tan Tan tốt trong nước, tan trong etanol
Định tính kali Đạt (Xem phương pháp thử).
| Khoảng nhiệt độ nóng chảy của acid sorbic dẫn xuất từ mẫu | 130 - 1350C |
Định tính liên kết chưa bão hòa Đạt (Xem phương pháp thử)
5.2. Độ tinh khiết
Giảm khối lượng khi làm khô Không lớn hơn 1% (1050C; 3 giờ)
Độ acid hoặc độ kiềm Không lớn hơn 1% (tính theo acid sorbic hoặc kali cacbonat - Xem phương pháp thử)
Aldehyd Không lớn hơn 0,1% (tính theo formaldehyd).
Chì Không lớn hơn 2 mg/kg.
5.3. Hàm lượng C6H7KO2 Không nhỏ hơn 98% C6H7KO2 (tính theo chế phẩm khan)
6. Phương pháp thử
6.1. Thử định tính
Thử định tính kali
- Các hợp chất chứa kali thường cho ngọn lửa màu tím. Trong trường hợp mẫu có vi lượng natri màu ngọn lửa của kali sẽ bị che phủ bởi ngọn lửa của natri.
- Pha dung dịch mẫu thử có nồng độ đặc, cho vào ống nghiệm. Trong môi trường trung tính, cho dung dịch natri bitartrat TT.221 vào ống nghiệm chứa dung dịch mẫu đã chuẩn bị, trong dung dịch phải tạo kết tủa tinh thể trắng, tốc độ kết tủa chậm. Tinh thể này tan được trong môi trường dung dịch amoniac TT.009, natri hydroxid hoặc carbonat. Có thể thúc đẩy tốc độ kết tinh bằng cách khuấy dung dịch trong ống nghiệm bằng đũa thủy tinh thêm bi thủy tinh, hoặc thêm lượng nhỏ acid acetic băng hoặc etanol.
Xác định khoảng nhiệt độ nóng chảy
Xem hướng dẫn chung các phương pháp thử (P.VL.01).
Ghi chú:
Dẫn xuất hóa acid sorbic từ mẫu thử; Acid hóa dung dịch mẫu thử bằng dung dịch acid hydrocloric loãng TT.127. Lọc thu kết tủa acid sorbic. Rửa kết tủa sạch Clo bằng nước cất. Làm khan kết tủa thu được trong chân không trên H2SO4.
Thử định tính liên kết chưa bão hòa
Pha dung dịch mẫu thử (1/10). Lấy 2ml dung dịch mẫu thử lắc với vài giọt dung dịch brom (nước brom) TT.048, màu nâu của nước brom sẽ mất.
6.2. Thử độ tinh khiết
Xác định giảm khối lượng khi làm khô
Xem hướng dẫn chung các phương pháp thử (P.VC.01).
Chú ý: Sấy ở nhiệt độ 1050C trong 3 giờ.
Xác định độ acid hoặc độ kiềm
- Hòa tan 1,1g mẫu vào trong 20ml nước cất và thêm 3 giọt chỉ thị là dung dịch phenolphtalein TT.179.
- Nếu dung dịch không màu, tiến hành chuẩn độ bằng dung dịch NaOH 0,1N đến khi xuất hiện màu hồng bền vững trong 15 giây. Lượng NaOH 0,1N sử dụng không quá 1,1ml.
- Nếu dung dịch có màu hồng, tiến hành chuẩn độ bằng dung dịch HCl 0,1N. Thể tích dung dịch HCl 0,1N sử dụng không quá 0,8ml.
Xác định hàm lượng aldehyd
- Chuẩn bị dung dịch mẫu thử 0,3%, điều chỉnh pH của dung dịch thử về 4 bằng HCl 1N, lọc dung dịch.
- Ống thử: Lấy 5ml dung dịch mẫu thử cho vào ống nghiệm và thêm vào đó 2,5ml dung dịch thuốc thử Schiff TT.211, để yên từ 10 đến 15 phút.
- Ống chuẩn: Lấy 5ml dung dịch formaldehyd có nồng độ 3μg/ml cho vào ống nghiệm, thêm vào đó 2,5ml dung dịch thuốc thử Schiff TT.211, để yên trong 10 đến 15 phút.
- Màu của ống thử không được đậm hơn màu của ống chuẩn.
Xác định hàm lượng chì
Xem hướng dẫn chung các phương pháp thử (P.VC.09).
6.3. Định lượng C6H7KO2
Cân chính xác 0,25g mẫu thử kali sorbat đã được làm khô. Hòa tan vào 36ml acid acetic băng và 4ml anhydrid acetic trong bình thủy tinh có nút 250ml, làm ấm dung dịch, để nguội về nhiệt độ phòng, thêm 2 giọt dung dịch metyl tím TT.265 và chuẩn độ với dung dịch acid percloric 0,1N trong acid acetic băng tới khi dung dịch chuẩn độ có màu xanh bền ít nhất 30 giây.Thao tác tương tự với mẫu trắng để xác định hệ số điều chỉnh, 1ml acid percloric 0,1N tương đương với 15,02mg C6H7KO2
QUY ĐỊNH ĐÁNH GIÁ VỆ SINH AN TOÀN CALCI SORBAT
1. Tên khác, ký hiệu Chỉ số INS: 203; Chỉ số EEC: E203
2. Định danh
Tên hóa học Calci sorbat, muối calci của tran, tran 2,4-hexadienoic acid.
Mã số C.A.S. 7492-55-9
Công thức hóa học C12H14CaO4
| Công thức phân tử |
|
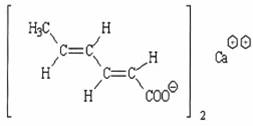
Khối lượng phân tử 262,32
3. Mô tả ngoại quan Bột tinh thể trắng mịn
4. Chức năng Chất bảo quản
5. Yêu cầu về vệ sinh an toàn
5.1. Định tính
Tính tan Tan trong nước, hầu như không tan trong etanol
Định tính calci Đạt (mô tả trong phần phương pháp thử)
| Khoảng nhiệt độ nóng chảy của acid sorbic dẫn xuất hóa từ mẫu | Từ 132 - 1350C |
Định tính liên kết chưa bão hòa Đạt (mô tả trong phần phương pháp thử).
5.2. Độ tinh khiết
Giảm khối lượng khi làm khô Không lớn hơn 3% (trên H2SO4 trong chân không, 4 giờ).
Flo Không lớn hơn 10mg/kg.
Aldehyd Không lớn hơn 0,1% (tính theo formaldehyd)
Chì Không lớn hơn 2mg/kg.
5.3. Định lượng C12H14O4Ca Không nhỏ hơn 98% và 102% C12H14CaO4 (tính theo chế phẩm khan)
6. Phương pháp thử
6.1. Thử định tính
Thử định tính Calci
Hoà tan mẫu thử vào nước cất (tỷ lệ 1/20), cho 2 giọt chỉ thị là dung dịch metyl đỏ TT.160, trung hòa dung dịch này bằng dung dịch amoniac TT.009, nhỏ giọt dung dịch amoni oxalat TT.023 vào dung dịch thử, trong dung dịch thử phải có kết tủa trắng. Kết tủa này không tan trong nước, acid acetic nhưng tan trong acid hydrocloric.
Muối của calci tẩm ướt bằng acid hydrocloric tạo ngọn lửa màu đỏ ánh vàng khi đốt bằng ngọn lửa đèn khí
Xác định khoảng nhiệt độ nóng chảy
Xem hướng dẫn chung các phương pháp thử (P.VL.01).
Ghi chú:
Dẫn xuất hóa acid sorbic từ mẫu thử: Acid hóa dung dịch mẫu thử bằng dung dịch acid hydrocloric loãng TT.127. Lọc thu kết tủa acid sorbic. Rửa kết tủa sạch Clo bằng nước cất. Làm khan kết tủa thu được trong chân không trên H2SO4.
Thử định tính liên kết chưa bão hòa.
Lắc khoảng 0,02g mẫu với 1ml dung dịch brom (nước brom) TT.048, màu nâu của nước brom sẽ mất.
6.2. Thử độ tinh khiết
Xác định giảm khối lượng khi làm khô
Làm khô trong chân không, trên H2SO4 đặc trong 4 giờ.
Xác định flo
Xem hướng dẫn chung các phương pháp thử (P.VC.06).
Ghi chú: Lấy 5g mẫu thử.
Xác định aldehyd
- Chuẩn bị dung dịch mẫu thử 0,3%, điều chỉnh pH của dung dịch thử về 4 bằng HCl 1N, lọc dung dịch.
- Ống thử: Lấy 5ml dung dịch mẫu thử cho vào ống nghiệm và thêm vào đó 2,5ml dung dịch thuốc thử Schiff TT.211, để yên từ 10 đến 15 phút.
- Ống chuẩn: Lấy 5ml dung dịch formaldehyd có nồng độ 3μg/ml cho vào ống nghiệm, thêm vào đó 2,5ml dung dịch thuốc thử Schiff TT.211, để yên trong 10 đến 15 phút.
- Màu của ống thử không được đậm hơn màu của ống chuẩn.
Xác định chì
Xem hướng dẫn chung các phương pháp thử (P.VC.09).
6.3. Định lượng C12H14O4Ca
Tiến hành:
Cân chính xác 0,25g mẫu thử calci sorbat đã được làm khô. Hòa tan vào trong 35ml acid acetic băng và 4ml anhydrid acetic trong bình thủy tinh có nút 250ml, làm ấm dung dịch, để nguội về nhiệt độ phòng, thêm 2 giọt dung dịch metyl tím TT.265 và chuẩn độ bằng dung dịch acid percloric 0,1N trong acid acetic băng tới khi dung dịch chuẩn độ có màu xanh bền ít nhất 30 giây. Thao tác tương tự với mẫu trắng để xác định hệ số điều hành, 1ml acid percloric 0,1N tương đương với 13,12mg C12H14CaO4.
QUY ĐỊNH ĐÁNH GIÁ VỆ SINH AN TOÀN NATRI BENZOAT
1. Tên khác, ký hiệu Chỉ số INS: 211; Chỉ số EEC: E211
2. Định danh
Tên hóa học Natri benzoat, muối natri của acid benzencacboxylic; muối natri của acid phenylcacboxylic.
Mã số C.A.S 532-32-1
Công thức hóa học C7H5NaO2
| Công thức phân tử |
|
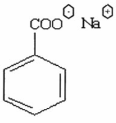
Khối lượng phân tử 144,11
3. Mô tả ngoại quan Bột tinh thể màu trắng hoặc dạng hạt nhỏ, hầu như không mùi
4. Chức năng Chất bảo quản
5. Yêu cầu về vệ sinh an toàn
5.1. Định tính
Tính tan Tan tốt trong nước, ít tan trong etanol
Định tính gốc benzoat Đạt (Xem phương pháp thử)
Định tính natri Đạt (Xem phương pháp thử)
5.2. Độ tinh khiết
Tính acid hoặc tính kiềm Đạt (xem phương pháp thử)
Giảm khối lượng khi làm khô Không lớn hơn 1,5% (1050C, 4 giờ)
Tổng kim loại nặng Không lớn hơn 10mg/kg
Các hợp chất dễ cacbon hóa Đạt (xem phương pháp thử)
Các hợp chất hữu cơ cơ clo Không lớn hơn 0,07%
Các hợp chất dễ oxy hóa Đạt (xem phương pháp thử)
5.3. Định lượng C7H5NaO2 Không nhỏ hơn 99% C7H5NaO2 (tính theo chế phẩm khan)
6. Phương pháp thử
6.1. Thử định tính
Thử định tính gốc benzoat
Dịch thử: Cân 1g mẫu pha với 10ml nước cất.
Thêm dung dịch sắt (III) clorid TT.112 vào dung dịch thử. Trong dung dịch thử phải xuất hiện kết tủa màu da bò.
Thử định tính natri
Acid hóa dung dịch mẫu bằng HNO3 loãng, cho dung dịch Coban uranyl acetat TT.071, lắc nhẹ trong vài phút. Trong dung dịch thử phải xuất hiện kết tủa màu vàng.
Các hợp chất chứa natri cho ngọn lửa màu vàng khi đốt bằng ngọn lửa đèn khí.
6.2. Thử độ tinh khiết
Xác định độ acid (hoặc độ kiềm)
Hòa tan 2g mẫu (cân chính xác đến mg) vào 20ml nước cất vừa đun sôi.
Chuẩn độ dung dịch này bằng NaOH 0,1N (hoặc HCl 0,1N) dùng phenolphatein (TT) làm chất chỉ thị màu.
Thể tích NaOH 0,1N (hoặc HCl 0,1N) dùng để chuẩn độ không được lớn hơn 0,5ml
Xác định giảm khối lượng khi làm khô
Xem hướng dẫn chung các phương pháp thử (P.VC.01)
Ghi chú: Sấy ở nhiệt độ 1050C trong 4 giờ.
Xác định tổng các kim loại nặng
Xem hướng dẫn chung các phương pháp thử (P.VC.10 - Phương pháp 1)
Chú ý: Cân 4g mẫu thử, hòa tan trong 40ml nước cất. Thêm từng giọt 10ml dung dịch acid hydrocloric loãng TT.127, vừa thêm vừa khuấy đều, lọc. Lấy 25ml dịch lọc tiến hành thử nghiệm.
Xác định các hợp chất dễ cacbon hóa
Hoà tan 0,5g mẫu (cân chính xác đến mg) vào 5ml dung dịch acid sulfuric TT.245, dung dịch có màu hồng. So sánh cường độ màu của dung dịch thử với cường độ màu của dung dịch đối chứng “Q" (xem hướng dẫn chung các phương pháp thử). Cường độ màu của dung dịch thử không được đậm hơn cường độ màu của dung dịch đối chứng “Q". Xem Hướng dẫn chung các phương pháp thử (P.HC.02).
Xác định các hợp chất hữu cơ cơ clo.
Cân chính xác 0,25g mẫu, hòa tan vào trong 10ml nước cất. Acid hóa bằng HNO3 10%, lọc lấy kết tủa. Trộn kết tủa với 0,5g CaCO3, sấy khô hỗn hợp và nung. Cho vào cặn sau nung 20ml HNO3 10% khuấy kỹ và lọc. Trộn dịch lọc với 0,5m AgNO3 0,1N.
Độ đục của dung dịch này không được lớn hơn độ đục của hỗn hợp: 19,5ml nước cất + 0,5ml HCl 0,01N và 0,5ml AgNO3 0,1N.
Xem Hướng dẫn chung các phương pháp thử (P.HC.01)
Xác định các hợp chất dễ oxy hóa.
Thêm 1,5ml H2SO4 vào 100ml nước cất, đun sôi và thêm từng giọt KMnO4 0,1N cho tới khi màu hồng bền trong 30 giây. Hòa 1g mẫu (cân chính xác tới mg) vào dung dịch đã được đun nóng và chuẩn độ bằng KMnO4 0,1N tới khi màu hồng bền trong 15 giây. Thể tích dung dịch KMnO4 0,1N dùng chuẩn độ không lớn hơn 0,5ml.
6.3. Định lượng C7H5NaO2
Cân chính xác 3g mẫu đã sấy khô trong 4 giờ ở 1050C, chuyển vào bình nón 250ml. Thêm 50ml nước cất để hòa tan mẫu. Trung hòa dung dịch bằng HCl 0,1N với chỉ thị là dung dịch phenolphtalein TT.179 (nếu cần). Thêm 50ml ete và vài giọt dung dịch bromophenol xanh TT.057. Chuẩn độ với dung dịch HCl 0,5N lắc bình cho tới khi màu của chỉ thị bắt đầu thay đổi. Chiết lớp nước ở dưới vào bình khác, rửa lớp ete với 10ml nước cất. Gộp dịch rửa vào bình chứa lớp nước chiết. Thêm 20ml ete vào dịch này. Tiếp tục chuẩn độ bằng dung dịch HCl 0,5N và lắc liên tục.
1ml dung dịch HCl 0,5N dùng để chuẩn độ tương ứng với 72,05mg C7H5NaO2
QUY ĐỊNH ĐÁNH GIÁ VỆ SINH AN TOÀN KALI BENZOAT
1. Tên khác, ký hiệu Chỉ số INS: 212; Chỉ số EEC: E212, Kali benzoat
2. Định danh
Tên hóa học muối kali của acid benzencacboxylic, muối kali của acid phenylcacboxylic.
Mã số C.A.S. 582-25-2
Công thức hóa học C7H5KO2.3H2O
| Công thức phân tử |
|
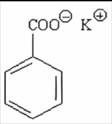
Khối lượng phân tử 214,27
3. Mô tả ngoại quan Dạng bột hoặc tinh thể màu trắng
4. Chức năng Chất bảo quản
5. Yêu cầu về vệ sinh an toàn
5.1. Định tính
Tính tan Tan tốt trong nước, tan trong etanol
Định tính gốc benzoat Đạt (mô tả trong phần phương pháp thử)
Định tính kali Đạt (mô tả trong phần phương pháp thử)
5.2. Độ tinh khiết
Giảm khối lượng khi làm khô Không lớn hơn 26,5% (1050C, 4h)
Độ acid và độ kiềm Đạt (mô tả trong phần phương pháp thử)
Tổng các kim loại nặng Không lớn hơn 10mg/kg.
Các hợp chất dễ cacbon hóa Đạt (mô tả trong phần phương pháp thử)
Các hợp chất dễ oxy hóa Đạt (mô tả trong phần phương pháp thử)
Các hợp chất hữu cơ clo Không lớn hơn 0,07% (tính theo Cl)
5.3. Định lượng C7H5KO2 Không nhỏ hơn 99% C7H5KO2 tính theo chế phẩm khan
6. Phương pháp thử
6.1. Thử định tính
Thử định tính kali
- Các hợp chất chứa kali thường cho ngọn lửa màu tím. Trong trường hợp mẫu có vi lượng natri màu ngọn lửa của kali sẽ bị che phủ bởi ngọn lửa của natri.
- Pha dung dịch mẫu thử có nồng độ 10%, cho vào ống nghiệm. Trong môi trường trung tính, cho dung dịch natri bitartrat TT.221 vào ống nghiệm chứa dung dịch mẫu đã chuẩn bị, trong dung dịch phải tạo kết tủa tinh thể trắng, tốc độ kết tủa chậm. Tinh thể này tan được trong môi trường dung dịch amoniac TT.009, natri hydroxid hoặc carbonat. Có thể thúc đẩy tốc độ kết tinh bằng cách khuấy dung dịch trong ống nghiệm bằng đũa thuỷ tinh thêm bi thuỷ tinh, hoặc thêm lượng nhỏ acid acetic băng hoặc etanol
Thử định tính gốc benzoat
Dịch thử: Cân 1g mẫu pha với 10ml nước cất
Thêm dung dịch sắt (III) clorid TT.112 vào dung dịch thử. Trong dung dịch thử phải xuất hiện kết tủa màu da bò.
6.2. Thử độ tinh khiết
Xác định độ acid và độ kiềm
Hòa tan 2g mẫu (cân chính xác đến mg) vào 20ml nước cất vừa đun sôi
Chuẩn độ dung dịch này bằng NaOH 0,1N (hoặc HCl 0,1N) dùng dung dịch phenolphtalein TT.179 làm chất chỉ thị màu.
Lượng NaOH 0,1N (hoặc HCl 0,1N) dùng để chuẩn độ không được lớn hơn 0,5ml.
Xác định tổng các kim loại nặng
Xem hướng dẫn chung các phương pháp thử (P.VC.10 - phương pháp 1)
Chú ý: Cân 4g mẫu thử, hòa tan trong 40ml nước cất. Thêm từng giọt 10ml dung dịch acid hydrocloric loãng TT.127, vừa thêm vừa khuấy đều, lọc. Lấy 25ml dịch lọc tiến hành thử nghiệm.
Xác định các hợp chất dễ cácbon hóa.
Cân 0,5g mẫu (chính xác đến mg), hòa tan trong 5ml dung dịch acid sulfuric TT.245. Dung dịch thử có màu hồng, so sánh cường độ màu của dung dịch thử với cường độ màu của dung dịch đối chứng “Q" (Xem hướng dẫn chung các phương pháp thử), màu của dung dịch thử không được đậm hơn màu của dung dịch đối chứng “Q". Xem hướng dẫn chung các phương pháp thử (P.HC.02).
Xác định các hợp chất dễ oxi hóa
Thêm 1,5ml H2SO4 vào 100ml nước cất, đun sôi và thêm từng giọt KMnO4 0,1N cho tới khi màu hồng bền trong 30 giây. Hòa tan 1g mẫu (cân chính xác tới mg) vào dung dịch đã được đun nóng và chuẩn độ bằng KMnO4 0,1N tới khi màu hồng bền trong 15 giây. Lượng KMnO4 0,1N dùng chuẩn độ không lớn hơn 0,5ml
Xác định các hợp chất hữu cơ cơ clo
Xem hướng dẫn chung các phương pháp thử (P.HC.01).
Ghi chú: cân 0,25g mẫu thử. Sử dụng 0,5ml dung dịch HCl 0,01N làm dung dịch chuẩn so sánh.
6.3. Định lượng C7H5KO2
Cân chính xác 3g mẫu đã sấy khô trong 4 giờ ở 1050C, chuyển vào bình nón 250ml. Thêm 50ml nước cất để hòa tan mẫu. Trung hòa dung dịch bằng HCl 0,1N với chỉ thị là dung dịch phenolphtalein TT.179. Thêm 50ml ete và vài giọt dung dịch bromophenol xanh TT.057. Chuẩn độ với acid HCl 0,5N lắc bình cho tới khi màu của chỉ thị bắt đầu thay đổi. Chiết lớp nước ở dưới vào bình khác, rửa lớp ete với 10ml nước cất. Gộp dịch rửa vào bình chứa lớp nước chiết. Thêm 20ml ete vào dịch này. Tiếp tục chuẩn độ với HCl 0,5N và lắc liên tục.
1ml HCl 05N dùng để chuẩn độ tương ứng với 80,11 mg C7H5KO2.
QUY ĐỊNH ĐÁNH GIÁ VỆ SINH AN TOÀN ETYL P - HYDROXYBENZOAT
1. Tên khác, ký hiệu Etylparaben, etyl p- hydroxybenzoat. Chỉ số INS: 214; Chỉ số EEC: E218
2. Định danh
Tên hóa học Etyl p - hydroxy benzoat, Etyl este của acid p - hydroxybenzoic.
Mã số C.A.S. 120-47-8
Công thức hoá học C9H10O3
| Công thức phân tử |
|
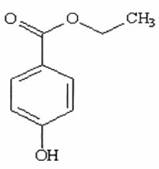
Khối lượng phân tử 166,18
3. Mô tả ngoại quan Bột tinh thể nhỏ, không mùi, không màu hoặc màu trắng
4. Chức năng Chất bảo quản
5. Yêu cầu về vệ sinh an toàn
5.1. Định tính
Tính tan Tan trong etanol, ete và propylen glycol
Khoảng nhiệt độ nóng chảy 115 - 1180C
Định tính p-hydroxybenzoat Đạt (mô tả trong phần phương pháp thử)
5.2. Độ tinh khiết
Giảm khối lượng khi làm khô Không lớn hơn 0,5% (800C; 2 giờ)
Tro sulfat Không lớn hơn 0,05%.
Độ acid Đạt (mô tả trong phần phương pháp thử)
| Acid p-hydroxybenzoic và acid salicylic | Đạt (mô tả trong phần phương pháp thử) |
Chì Không lớn hơn 2mg/kg.
5.3. Định lượng C9H10O3 Không nhỏ hơn 99% C9H10O3 (tính theo chế phẩm khan)
6. Phương pháp thử
6.1. Thử định tính
Xác định khoảng nóng chảy
Xem hướng dẫn chung các phương pháp thử (P.VL.01).
Thử định tính p-hydroxybenzoat
Cân 0,5g mẫu thử thêm vào 10ml dung dịch natri hydroxid TT.230, đun sôi 30 phút và cô đặc đến còn khoảng 5ml, để nguội, acid hóa bằng dung dịch acid sulfuric loãng TT.246. Lọc lấy kết tủa, rửa kỹ kết tủa bằng nước cất. Làm khô trong bình hút ẩm với H2SO4 đặc. Xác định khoảng nhiệt độ nóng chảy của kết tủa thu được (acid p-hydroxybenzoic).
Khoảng nhiệt độ nóng chảy phải đạt: 2120C - 2170C
6.2. Thử độ tinh khiết
Giảm khối lượng khi làm khô
Xem hướng dẫn chung các phương pháp thử (P.VC.01)
Ghi chú: Sấy ở 800C, trong 2 giờ.
Xác định tro sulfat
Xem hướng dẫn chung các phương pháp thử (P.HC.11-Phương pháp 1)
Chú ý: Cân 2g mẫu thử
Xác định độ acid
Đun nóng 750mg mẫu thử với 15ml nước cất ở 800C trong 1 phút, để nguội và lọc. Thử dịch lọc bằng giấy quỳ, pH dịch lọc có thể acid hoặc trung tính. Lấy 10ml dịch lọc, thêm 0,2ml NaOH 0,1N và 2 giọt dung dịch metyl đỏ TT.160. Dung dịch phải có màu vàng (không có màu hồng).
Xác định acid p-hydroxybenzoic và acid salicilic
Hòa tan 0,5g mẫu thử vào trong 30ml ete, thêm 20ml dung dịch NaHCO3 1%, lắc đều, tách lấy lớp nước. Rửa lớp nước hai lần bằng ete (mỗi lần 20ml), thêm 5ml dung dịch acid sulfuric loãng TT.246 và 30ml ete, lắc kỹ. Tách lấy lớp ete, thêm 10ml nước cất và lắc kỹ. Lọc lớp ete, tráng rửa bình và màng lọc bằng một ít ete. Gộp dịch rửa và dịch lọc (dịch ete), cất quay loại ete trong nồi cách thủy, làm khô cặn thu được trên H2SO4 đặc tới khối lượng không đổi. Khối lượng của cặn không được quá 5mg.
Hoà tan cặn này trong 25ml nước cất, đun nóng tới khoảng 7000C, lọc và nhỏ vài giọt dung dịch sắt (III) clorid loãng TT.114. Dung dịch mẫu thử phải chuyển thành màu tím đỏ.
Xác định chì
Xem hướng dẫn chung các phương pháp thử (P.HC.09)
6.3. Định lượng C9H10O3
Dung dịch đệm pH =6,5:
Trộn 50ml dung dịch kali dihydro phosphat 0,2M với 15,2ml dung dịch natri hydroxid 0,2M. Định mức đến 200ml bằng nước cất.
Tiến hành:
Cân chính xác 2g mẫu thử khan và chuyển vào bình thủy tinh. Thêm 40ml NaOH 1N và rửa thành bình bằng nước cất. Đậy mặt kính hoặc nắp hộp lồng, đun sôi dung dịch trong 1 giờ và để nguội. Thêm 5 giọt dung dịch bromothymol xanh TT.060 và chuẩn độ lượng NaOH dư bằng H2SO4 1N, so sánh màu của dung dịch thu được với màu của dung dịch đệm pH = 6,5 (TT) có chứa cùng tỷ lệ chất chỉ thị.
Tiến hành tương tự với mẫu trắng để xác định hệ số hiệu chỉnh (nếu cần).
1ml NaOH 1N tương đương với 166,18mg C9H10O3.
QUY ĐỊNH ĐÁNH GIÁ VỆ SINH AN TOÀN PROPYL P-HYDROXYBENZOAT
1. Tên khác, ký hiệu Propylparaben, propyl p-hydroxybenzoat, Chỉ số INS: 216; Chỉ số EEC: E216
2. Định danh
Tên hóa học Propyl p-hydroxybenzoat, este của acid p-hydroxybenzoic và n-propyl
Mã số C.A.S 94-13-3
Công thức hóa học C10H12O3
| Công thức phân tử |
|
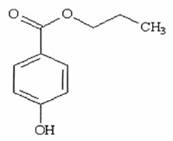
Khối lượng phân tử 180,21
3. Mô tả ngoại quan Tinh thể không màu, tinh thể nhỏ hoặc bột tinh thể màu trắng và hầu như không mùi
4. Chức năng Chất bảo quản
5. Yêu cầu về vệ sinh an toàn
5.1. Định tính
Tính tan Tan trong etanol, ete và propylen glycol
Khoảng nhiệt độ nóng chảy 950C đến 980C
Định tính p-hydroxybenzoat Đạt (mô tả trong phần phương pháp thử)
5.2. Độ tinh khiết
Giảm khối lượng khi làm khô Không lớn hơn 0,5% (trên silicagel; 5 giờ)
Tro sulfat Không lớn hơn 0,05%
Độ acid Đạt (mô tả trong phần phương pháp thử)
| Acid p-hydroxybenzoic và acid salisilic | Không lớn hơn 10g/kg |
Chì Không lớn hơn 2mg/kg
5.3. Định lượng C10H12O3 Không nhỏ hơn 99% C10H12O3 (tính theo chế phẩm khan)
6. Phương pháp thử
6.1. Thử định tính
Xác định khoảng nhiệt độ nóng chảy
Xem hướng dẫn chung các phương pháp thử (P.VL.01)
Thử định tính p-hydroxybenzoat
Cân 0,5g mẫu thử thêm vào 10ml dung dịch natri hydroxid TT.230, đun sôi 30 phút và cô đặc đến còn khoảng 5ml, để nguội, acid hóa bằng dung dịch acid sulfuric loãng TT.246. Lọc lấy kết tủa, rửa kỹ kết tủa bằng nước cất. Làm khô trong bình hút ẩm với H2SO4 đặc. Xác định khoảng nhiệt độ nóng chảy của kết tủa thu được (acid p - hydroxybenzoic).
Khoảng nhiệt độ nóng chảy phải đạt: 2120C - 2170C.
6.2. Thử độ tinh khiết
Xác định giảm khối lượng khi làm khô
Xem hướng dẫn chung các phương pháp thử (P.VC.01)
Ghi chú: Mẫu được làm khô bằng Silicagel trong 4 giờ
Xác định tro sulfat
Xem hướng dẫn chung các phương pháp thử (P.VC.11-Phương pháp 2)
Chú ý: Lấy 2g mẫu thử
Xác định độ acid
Đun nóng 750mg mẫu thử với 15ml nước cất ở 800C trong 1 phút, để nguội và lọc. Thử dịch lọc bằng giấy quỳ, pH dịch lọc có thể acid hoặc trung tính. Lấy 10ml dịch lọc, thêm 0,2ml NaOH 0,1N và 2 giọt dung dịch metyl đỏ TT.160. Dung dịch phải có màu vàng (không có màu hồng).
Xác định acid p-hydroxybenzoic và acid salicylic
Hòa tan 0,5g mẫu thử vào trong 30ml ete, thêm 20ml dung dịch NaHCO3 1%, lắc đều, tách lấy lớp nước. Rửa lớp nước hai lần bằng ete (mỗi lần 20ml), thêm 5ml dung dịch acid sulfuric loãng TT.246 và 30ml ete, lắc kỹ. Tách lấy lớp ete, thêm 10ml nước cất và lắc kỹ. Lọc lớp ete, tráng rửa bình và màng lọc bằng một ít ete. Gộp dịch rửa và dich lọc (dịch ete), cất quay loại ete trong nồi cách thuỷ, làm khô cặn thu được trên H2SO4 đặc tới khối lượng không đổi. Khối lượng của cặn không được quá 5mg
Hòa tan cặn này trong 25ml nước cất, đun nóng tới khoảng 7000C, lọc và nhỏ vài giọt dung dịch sắt (III) clorid loãng TT.114. Dung dịch mẫu thử phải chuyển thành màu tím đỏ.
Xác định chì
Xem hướng dẫn chung các phương pháp thử (P.VC.09)
6.3. Định lượng C10H12O3
Dung dịch đệm pH = 6,5:
Trộn 50ml dung dịch kali dihydro phosphat 0,2M với 15,2ml dung dịch natri hydroxid 0,2M. Định mức đến 200ml bằng nước cất.
Tiến hành:
Cân chính xác 2g mẫu thử khan và chuyển vào trong bình thủy tinh. Thêm 40ml NaOH 1N và rửa thành bình bằng nước cất. Đậy nắp (mặt kính hoặc nắp hộp lồng), đun sôi dung dịch trong 1 giờ và để nguội. Thêm 5 giọt dung dịch bromothymol xanh TT.060 và chuẩn độ lượng NaOH dư bằng H2SO4 1N, so sánh màu của dung dịch thu được với màu của dung dịch đệm pH = 6,5 (TT) có chứa cùng tỷ lệ chất chỉ thị.
Tiến hành xác định mẫu trắng với thuốc thử và điều chỉnh nếu cần thiết.
1ml NaOH 1N tương đương với 180,2mg C10H12O3.
QUY ĐỊNH ĐÁNH GIÁ VỆ SINH AN TOÀN METYL P - HYDROXYBENZOAT.
1. Tên khác, ký hiệu Metyl p-hydroxybenzoat, Metyl paraben, metyl p - oxybenzoat. Chỉ số INS: 218; Chỉ số EEC: E218
2. Định danh
Tên hóa học Metyl p-hydroxybenzoat, metyl este của acid p-hydroxybenzoic
Mã số C.A.S 99-76-3
Công thức hóa học C8H8O3
| Công thức phân tử |
|
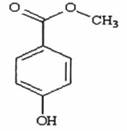
Khối lượng phân tử 152,15
3. Mô tả ngoại quan Tinh thể nhỏ hầu như không mùi, không màu hoặc bột tinh thể màu trắng
4. Chức năng Chất bảo quản
5. Yêu cầu về vệ sinh an toàn
5.1. Định tính
Tính tan Tan trong etanol, ete; ít tan trong nước và propyl glycol
Khoảng nhiệt độ nóng chảy 125 đến 1280C
Định tính p-hydroxybenzoat Đạt (mô tả trong phần phương pháp thử)
5.2. Độ tinh khiết
Giảm khối lượng khi làm khô Không lớn hơn 0,5% (trên silicagel; 5 giờ)
Tro sulfat Không lớn hơn 0,05%
Độ acid Đạt (mô tả trong phần phương pháp thử)
| Acid p-hydroxybenzoic và acid salicilic | Đạt (mô tả trong phần phương pháp thử) |
Chì Không lớn hơn 2mg/kg
5.3. Định lượng C8H8O3 Không nhỏ hơn 99% C8H8O3 (tính theo chế phẩm khan)
6. Phương pháp thử
6.1. Thử định tính
Xác định khoảng nhiệt độ nóng chảy
Xem hướng dẫn chung các phương pháp thử (P.VL.01)
Thử định tính p-hydroxybenzoat
Cân 0,5g mẫu thử thêm vào 10ml dung dịch natri hydroxid TT.230, đun sôi 30 phút và cô đặc đến còn khoảng 5ml, để nguội, acid hóa bằng dung dịch acid sulfuric loãng TT.246. Lọc lấy kết tủa, rửa kỹ kết tủa bằng nước cất. Làm khô trong bình hút ẩm với H2SO4 đặc. Xác định khoảng nhiệt độ nóng chảy của kết tủa thu được (acid p - hydroxybenzoic).
Khoảng nhiệt độ nóng chảy phải đạt: 2120C - 2170C.
6.2. Thử độ tinh khiết
Xác định giảm khối lượng khi làm khô
Xem hướng dẫn chung các phương pháp thử (P.VC.01)
Mẫu được làm khô bằng Silicagel trong 5 giờ
Xác định tro sulfat
Xem hướng dẫn chung các phương pháp thử (P.HC.11-Phương pháp 1)
Chú ý: Lấy 2g mẫu thử
Xác định độ acid
Đun nóng 750mg mẫu thử với 15ml nước cất ở 800C trong 1 phút, để nguội và lọc. Thử dịch lọc bằng giấy quỳ, pH dịch lọc có thể acid hoặc trung tính. Lấy 10ml dịch lọc, thêm 0,2ml NaOH 0,1N và 2 giọt dung dịch metyl đỏ TT.160. Dung dịch phải có màu vàng (không có ánh hồng).
Xác định acid p-hydroxybenzoic và acid salicilic
Hòa tan 0,5g mẫu thử vào trong 30ml ete, thêm 20ml dung dịch NaHCO3 1%, lắc đều, tách lấy lớp nước. Rửa lớp nước hai lần bằng ete (mỗi lần 20ml), thêm 5ml dung dịch acid sulfuric loãng TT.246 và 30ml ete, lắc kỹ. Tách lấy lớp ete, thêm 10ml nước cất và lắc kỹ. Lọc lớp ete, tráng rửa bình và màng lọc bằng một ít ete. Gộp dịch rửa và dich lọc (dịch ete), cất quay loại ete trong nồi cách thuỷ, làm khô cặn thu được trên H2SO4 đặc tới khối lượng không đổi. Khối lượng của cặn không được quá 5mg
Hòa tan cặn này trong 25ml nước cất, đun nóng tới khoảng 700C, lọc và nhỏ vài giọt dung dịch sắt (III) clorid loãng TT.114. Dung dịch mẫu thử phải chuyển thành màu tím đỏ.
Xác định chì
Xem hướng dẫn chung các phương pháp thử (P.HC.09)
6.3. Định lượng C8H8O3
Dung dịch đệm pH = 6,5:
Trộn 50ml dung dịch kali dihydro phosphat 0,2M với 15,2ml dung dịch natri hydroxid 0,2M. Định mức đến 200ml bằng nước cất.
Tiến hành:
Cân chính xác 2g mẫu thử khan và chuyển vào trong bình thủy tinh. Thêm 40ml NaOH 1N và rửa thành bình bằng nước cất. Đậy mặt kính hoặc nắp hộp lồng, đun sôi dung dịch trong 1 giờ và để nguội. Thêm 5 giọt dung dịch bromothymol xanh TT.060 và chuẩn độ lượng NaOH dư bằng H2SO4 1N, so sánh màu của dung dịch thu được với màu của dung dịch đệm pH = 6,5 (TT) có chứa cùng tỷ lệ chất chỉ thị.
Tiến hành tương tự với mẫu trắng để xác định hệ số điều chỉnh (nếu cần).
1ml NaOH 1N tương đương với 152,2mg C8H8O3.
QUY ĐỊNH ĐÁNH GIÁ VỆ SINH AN TOÀN KALI NITRAT
1. Tên khác, ký hiệu INS No. 252; EEC No. E252
2. Định danh
Tên hóa học Kali nitrat
Mã số C.A.S. 7757-79-1
Công thức hóa học KNO3
Công thức phân tử KNO3
Khối lượng phân tử 101.11
3. Mô tả ngoại quan Không mùi, không màu, tinh thể hình trụ trong suốt hoặc tinh thể trắng
4. Chức năng Chất bảo quản, chất ổn định màu
5. Yêu cầu về vệ sinh an toàn
5.1. Định tính
Tính tan Tan trong nước, ít tan trong etanol
Định tính kali Đạt (xem phương pháp thử)
Định tính gốc nitrat Đạt (xem phương pháp thử)
5.2. Độ tinh khiết
Giảm khối lượng khi làm khô Không lớn hơn 1% (1050C; 4 giờ)
Nitrit Không lớn hơn 20mg/kg
Asen Không lớn hơn 3mg/kg
Chì Không lớn hơn 10mg/kg
Tổng các kim loại nặng Không lớn hơn 20mg/kg
5.3. Định lượng KNO3 Không nhỏ hơn 99% KNO3 (tính theo chế phẩm khan)
6. Phương pháp thử
6.1. Thử định tính
Thử định tính kali
- Các hợp chất chứa kali thường cho ngọn lửa màu tím. Trong trường hợp mẫu có vi lượng natri màu ngọn lửa của kali sẽ bị che phủ bởi ngọn lửa của natri.
- Pha dung dịch mẫu thử có nồng độ đặc, cho vào ống nghiệm. Trong môi trường trung tính, cho dung dịch natri bitartrat TT.221 vào ống nghiệm chứa dung dịch mẫu đã chuẩn bị, trong dung dịch phải tạo kết tủa tinh thể trắng, tốc độ kết tủa chậm. Tinh thể này tan được trong môi trường dung dịch amoniac TT.009, natri hydroxid hoặc carbonat. Có thể thúc đẩy tốc độ kết tinh bằng cách khuấy dung dịch trong ống nghiệm bằng đũa thủy tinh thêm bi thủy tinh, hoặc thêm lượng nhỏ acid acetic băng hoặc etanol.
Thử định tính gốc nitrat.
Hóa chất:
- Thuốc thử sắt (II) sulfat: hoà tan 8g tinh thể (FeSO4.7H2O) trong 100ml nước cất. Chuẩn bị dung dịch trước khi dùng.
Tiến hành:
Thêm vào 3ml dung dịch mẫu thử 3ml axit sunfuric đặc, để nguội, trộn đều. Thêm từ từ dung dịch thuốc thử sắt (II) sulfat, tại ranh giới tiếp xúc của 2 dung dịch phải xuất hiện màu nâu.
6.2. Thử độ tinh khiết
Xác định giảm khối lượng khi làm khô
Xem Hướng dẫn chung các phương pháp thử (P.VC.01)
Ghi chú: Mẫu được sấy ở 1050C trong 4 giờ.
Xác định nitrit
Nguyên tắc:
Nitrit phản ứng với sulphanilamit và N-(1-naphtyl) etylendiamin dihydroclorid tạo phức có màu hồng. Xác định hàm lượng nitrit dựa trên độ hấp thụ quang của phức này tại bước sóng 540nm.
Hóa chất, thuốc thử: Tất cả các hóa chất thuốc có chất lượng tinh khiết phân tích
- Dung dịch sulphanilamit: hoà tan 2g sulphanilamit vào 1000ml dung dịch acid hydrloric loãng TT.127.
- Thuốc thử tạo phức: hoà tan 2g N-(1-naphtyl) etylendiamin dihydroclorid trong nước cất, định mức đến 100ml. Giữ dung dịch trong chai màu và trong tủ lạnh.
- Chuẩn Nitrit:
Dung dịch chuẩn gốc (500μg NO2-/ml): Hoà tan 0,750g NaNO2 (đã được làm khô trong bình hút ẩm silicagel trong 4 giờ) vào nước cất, định mức đến 1000ml.
Dung dịch chuẩn trung gian (50μg NO2-/ml): Hút 10ml chuẩn gốc, định mức đến 100ml bằng nước cất.
Dung dịch chuẩn trung gian (0,5μg NO2-/ml): Hút 10ml chuẩn trung gian, định mức đến 1000ml bằng nước cất.
Xây dựng đường chuẩn:
Lần lượt hút 0; 5; 10; 20; 50ml chuẩn làm việc (tương ứng với 0; 2,5; 5; 10; 25 μg NO2-), cho vào các bình định mức 100ml. Thêm nước cất đến khoảng 80ml, thêm vào mỗi bình 10ml dung dịch sulphanilamit, lắc đều. Sau 3 phút thêm vào mỗi bình 1ml dung dịch thuốc thử tạo phức, định mức đến vạch bằng nước cất. Trộn đều và để yên trong 15 phút. Đo độ hấp thụ quan của dãy dung dịch chuẩn tại bước sóng 540nm (cuvet đo quang 10mm). Dựng đường chuẩn biểu thị mối tương quan giữa độ hấp thụ quang và lượng NO2- của dung dịch (đường chuẩn là đường thẳng).
Tiến hành:
Cân 1g (chính xác đến mg) mẫu thử. Hòa tan trong nước cất và định mức đến 100ml. Hút 20ml dung dịch này cho vào bình định mức 100ml, thêm nước cất tới khoảng 80ml, thêm 10ml dung dịch sulphanilamit, lắc đều. Sau 3 phút thêm 1ml dung dịch thuốc thử tạo phức, định mức đến vạch bằng nước cất. Trộn đều và để yên trong 15 phút. Đo độ hấp thụ quan của dãy dung dịch chuẩn tại bước sóng 540nm (cuvet đo quang 10nm). Dựa vào đường chuẩn tính lượng NO2- có trong dung dịch thử.
Tính kết quả:
Hàm lượng Nitrit (mg/kg) =
Trong đó:
mx: Lượng NO2- trong dịch thử (μg)
m: khối lượng mẫu thử
Xác định Asen
Xem hướng dẫn chung các phương pháp thử (P.VC.08)
Chú ý: hoà tan 1g mẫu vào 3ml nước cất, thêm 2ml H2SO4. Đun cho đến khi có khói SO3 bay ra mạnh. Làm mát và rửa thành bình bằng nước cất. Tiếp tục đun cho đến khi có khói bay ra. Tiếp tục làm như vậy 3 lần. Làm nguội bình và pha loãng tới 35ml. Lấy dịch này tiến hành thử nghiệm.
Xác định chì
Xem hướng dẫn chung các phương pháp thử (P.VC.07)
Chú ý:
Chuẩn bị dung dịch thử: hoà tan 1g mẫu vào 10ml
Dung dịch chuẩn Pb: chứa 10μg Pb2+
Xác định tổng các kim loại nặng
Xem hướng dẫn chung các phương pháp thử (P.VC.02-Phương pháp 1)
Chú ý: Dung dịch thử: hòa tan 2g mẫu vào 25ml nước cất
6.3. Định lượng KNO3
Cân chính xác 0,4g mẫu thử đã được làm khô ở 1050C trong 4 giờ và hòa tan với khoảng 300ml nước trong bình cầu thể tích 500ml. Thêm 3g bột hợp kim Devada và 15ml dung dịch NaOH 40% vào bình, nối bộ phận chống phun và sinh hàn vào bình cầu. Cho 50ml H2SO4 0,1N vào trong bình hứng, để yên trong 2 giờ. Chưng cất lấy 250ml dịch chưng cất. Chuẩn độ H2SO4 dư bằng NaOH 0,1N dùng 3 giọt dung dịch metyl đỏ/metylen xanh TT.161 làm chất chỉ thị. Tiến hành phân tích mẫu trắng như với mẫu thử để hiệu chỉnh kết quả. 1ml H2SO4 0,1N tương ứng với 10,11mg KNO3.
QUY ĐỊNH ĐÁNH GIÁ VỆ SINH AN TOÀN TERT-BUTYLHYDROQUINON
1. Tên khác, ký hiệu TBHQ, Chỉ số INS: 319
2. Định danh
Tên hóa học Mono-tert-butylhydroquinon; t-butylhydroquinon 2-(1,1-dimethylethyl)-1,4-benzenediol
Mã số C.A.S. 1948-33-0
Công thức hóa học C10H14O2
| Công thức phân tử |
|
Khối lượng phân tử 166,22
3. Mô tả ngoại quan Tinh thể màu trắng rắn và có mùi đặc trưng
4. Chức năng Chất chống oxy hóa
5. Yêu cầu về vệ sinh an toàn
5.1. Định tính
Tính tan Hầu như không tan trong nước, tan trong etanol
Khoảng nhiệt độ nóng chảy 126,50C - 128,50C
Định tính Phenol Đạt yêu cầu (Xem phương pháp thử)
5.2. Độ tinh khiết
Tổng các kim loại nặng Không lớn hơn 10mg/kg
t-butyl-p-benzoquinon Không lớn hơn 0,2%
2,5-di-t-butyl-hydroquinon Không lớn hơn 0,2%
Hydroquinon Không lớn hơn 0,1%
Toluen Không lớn hơn 25mg/kg
5.3. Định lượng C10H14O2 Không nhỏ hơn 99,0% C10H14O2
6. Phương pháp thử
6.1. Thử định tính
Xác định khoảng nhiệt độ nóng chảy
Xem hướng dẫn chung các phương pháp thử (P.VL.01)
Thử định tính Phenol
Hòa tan khoảng 5mg mẫu vào 10ml metanol sau đó thêm 10.5ml dung dịch dimetylamin (1/4). Dung dịch phải xuất hiện màu đỏ (hồng).
6.2. Thử độ tinh khiết
Xác định tổng các kim loại nặng
Xem hướng dẫn chung các phương pháp thử (P.VC.10-Phương pháp 2)
Chú ý: Lấy 2g mẫu thử
Xác định t-Butyl-p-benzoquinon
Thiết bị:
Sử dụng máy quang phổ hồng ngoại hai chùm tia. Cuvet có cửa sổ CaF2 với chiều dày lớp mẫu lỏng là 0,4mm.
Hóa chất và thuốc thử:
Chuẩn bị dung dịch chuẩn: Cân chính xác 10mg mono-tert-butyl-p-benzoquinon chuẩn và chuyển vào trong bình định mức 10ml, hòa tan trong cacbon tetraclorua, pha loãng và định mức đến 10ml bằng dung môi trên rồi trộn đều.
Chuẩn bị mẫu thử: Cân chính xác 1g mẫu đã được nghiền thành bột mịn trong máy xay cao tốc rồi cho vào bình định mức 10ml, hòa tan trong cacbon tetraclorua, pha loãng cho đủ 10ml bằng dung môi trên rồi trộn đều. Lọc dung dịch qua màng lọc (UHWPO 1300) hoặc tương đương trước khi tiến hành thử.
Tiến hành:
Cho CCl4 vào cuvet so sánh và cho dung dịch chuẩn vào cuvet đo mẫu thử. Đặt lần lượt các cuvet này vào vị trí đo của máy. Ghi lại phổ hồng ngoại từ 1600 đến 1775cm-1. Trên phổ, vẽ đường nền từ 1612 đến 1750cm-1 và xác định độ hấp thụ (đã trừ nền) A0 của dung dịch chuẩn tại bước sóng 1659cm-1, xác định độ hấp thụ của dung dịch mẫu tại bước sóng 1659cm-1.
Tính kết quả:
Z =
Trong đó:
Z: Hàm lượng t-butyl-p-benzoquinon trong mẫu thử (%)
W0: khối lượng thực của dung dịch mono tertiary-butyl-p benzoquinon chuẩn (mg).
W1: khối lượng thực của mẫu thử (mg)
A0: độ hấp thụ của dung dịch chuẩn
A1: độ hấp thụ của dung dịch mẫu
Xác định 2,5-Di-t-butyl hydroquinon và hydroxyquinon
Thiết bị:
Máy sắc ký khí có detertor dẫn nhiệt (loại 810 F, M hoặc tương đương) gồm cột thép không gỉ có kích thước 0,61m (2ft) x 6,35mm (đường kính ngoài). Chất nhồi cột là 20% silicon SE -30 và 80% là diatoport S (60/80mesh) hoặc các vật liệu tương đương.
• Điều kiện vận hành:
Các thông số vận hành có thể biến đổi tuỳ thuộc vào từng thiết bị. Tuy nhiên có thể sử dụng máy sắc ký với các điều kiện sau:
- Nhiệt độ cột: lập chương trình từ 1000C đến 2700C, với 150C/1 phút.
- Nhiệt độ buồng bơm mẫu: 3000C
- Khí mang: He, tốc độ dòng là 100ml/phút
- Dòng cầu đo: 140mA
- Độ nhạy: 1x đối với tích phân kế (Infotronics CRS-100), 2x cho máy ghi
Hóa chất và thuốc thử:
- Dung dịch gốc: cân chính xác 50mg hydroquinon (HQ), 50mg 2,5-di-t-butylhydroquinon (DTBHQ) và 50mg metyl benzoat (chất nội chuẩn) rồi chuyển vào 3 bình định mức 50ml riêng biệt, pha loãng cho đủ 50ml bằng pyridin và trộn đều.
- Dãy dung dịch chuẩn: cho 0,5ml; 1ml; 2ml và 3ml dung dịch gốc HQ vào 4 bình định mức 10ml riêng biệt, cho thêm 2ml dung dịch metyl benzoat (chất nội chuẩn) vào mỗi bình, pha loãng cho đủ 10ml bằng pyridin rồi trộn đều. Tương tự, chuẩn bị 4 dung dịch chuẩn DTBHQ. Chuẩn bị dẫn xuất trimetylsilyl cho mỗi dung dịch như sau: Thêm 9 giọt dung dịch chuẩn vào xilanh khí kích thước 2ml, cho thêm 250μl của N,O bis-trimethylsilyl-acetamit rồi đun nóng ở 800C trong 10 phút. Chạy sắc ký hai lần, mỗi lần 10μl của mỗi dung dịch chuẩn và vẽ đồ thị tỷ lệ nồng độ của HQ trên nồng độ chất nội chuẩn (trục X) dựa vào tỷ lệ đáp ứng của nồng độ HQ trên nồng độ chất nội chuẩn (trục Y). Tương tự, vẽ đồ thị biểu thị mối quan hệ giữa DBHQ và chất nội chuẩn.
Tiến hành thử:
Cân chính xác khoảng 1g mẫu rồi chuyển vào bình định mức 10ml, cho thêm 2ml dung dịch gốc nội chuẩn metyl benzoat, pha loãng cho đủ 10ml bằng pyridin rồi trộn đều. Chuẩn bị dẫn xuất trimetylsilyl giống như trong phần chuẩn bị dãy dung dịch chuẩn ở trên. Sau đó chạy sắc ký hai lần, bơm 10μl/mỗi lần để ghi phổ sắc ký. Thời gian lưu của pic xấp xỉ nhau: metyl benzoat: 2,5 phút; dẫn xuất TMS của HQ: 5,5 phút; dẫn xuất TMS của TBHQ: 7,3 phút; dẫn xuất TMS của DTBHQ: 8,4 phút
Tính kết quả:
Xác định diện tích pic (đáp ứng). Tính tỷ lệ diện tích pic của HQ và DTBHQ trên chất nội chuẩn
Từ đường chuẩn, xác định tỷ lệ nồng độ của HQ và DTBHQ trên chất nội chuẩn và tính % của HQ và % DTBHQ trong mẫu theo công thức:
Z =
Trong đó:
Z là % HQ hoặc DTBHQ trong mẫu
Y là tỷ lệ nồng độ (trục X trên đường chuẩn).
I là % (m/v) của chất nội chuẩn trong mẫu thử
M là khối lượng của mẫu thử (g).
Xác định toluen
Thiết bị:
Máy sắc ký khí có detertor ion hóa ngọn lửa (loại F, M 810 hoặc tương đương) gồm cột thép không gỉ có kích thước 3,66m (12ft) x 3,18mm (đường kính ngoài). Pha tĩnh là 10% sillicon SE - 30 và 90% diatoport S (60/80mesh) (theo trọng lượng) hoặc các vật liệu tương đương.
• Điều kiện vận hành
Các thông số vận hành có thể thay đổi tùy thuộc vào từng loại thiết bị, nhưng chủ yếu dùng máy sắc ký với các điều kiện sau:
- Nhiệt độ cột: chương trình nhiệt từ 700C đến 2800C với tốc độ gia nhiệt là 150C/phút và giữ yên.
- Nhiệt độ buồng bơm mẫu: 2750C.
- Nhiệt độ cell: 3000C
- Đặt H2 và O2: 1,4atm (20 Psi).
Hóa chất và thuốc thử:
- Dung dịch chuẩn: Chuẩn bị dung dịch toluen trong octanol chứa khoảng 50μg/ml. Tính nồng độ thực (Cr) (%, m/v)
- Dung dịch mẫu: cân chính xác 2g mẫu rồi chuyển vào trong bình định mức 10ml, hoà tan trong octanol, pha loãng cho đủ 10ml bằng dung môi trên và trộn đều. Tính nồng độ thực của dung dịch (Cs) (%, m/v)
Tiến hành:
Bơm 5μl dung dịch chuẩn vào trong máy sắc ký và đo chiều cao pic toluen (Hr) trên sắc đồ. Thời gian lưu của toluen là 3,3 phút. Những pic khác là không cần quan tâm đến trong phép phân tích này.
Tương tự bơm 5μl dung dịch chuẩn và 5μl mẫu trắng có octanol vào máy sắc ký rồi đo chiều cao pic của toluen (Hs).
Tính kết quả:
T = HS / CR x CR / CS x 106
6.3. Định lượng C10H14O2
Cân chính xác 170mg mẫu đã nghiền thành bột mịn, chuyển vào bình nón miệng rộng 250ml, hoà tan trong 10ml metanol. Cho thêm 150ml nước cất, 1ml H2SO4 1N và 4 giọt chỉ thị diphenylamin (3mg Natri p-diphenylamin sunfonat trong 1ml H2SO4 0,1N). Chuẩn độ bằng ceri sulfat 0,1N tới khi màu dung dịch chuyển từ màu vàng sang tím đỏ. Ghi lại thể tích của ceri sulfat 0,1N (V; ml).
Tính % của C10H14O2 trong mẫu, không hiệu chỉnh đối với hydroquinon (HQ) và 2,5 di-tert-butylhydro-quinon (DTBHQ) theo công thức sau:
![]()
Trong đó:
- 0,1ml: Thể tích của ceri sulfat tiêu hao do các sản phẩm oxy hóa sơ cấp của tert-butylhydroquinon có trong mẫu.
- N: nồng độ thực của dung dịch ceri sulfat
- W: khối lượng của mẫu lấy phân tích (g).
Ghi lại % chưa hiệu chỉnh được hiệu chỉnh là A. Nếu HQ và DTBHQ có mặt trong mẫu, chúng cũng tham gia vào phản ứng chuẩn độ. Tính % thực của C10H14O2 trong mẫu theo công thức sau:
% thực của C10H14O2 = A - (%HQ x 1,51) - (% DTBHQ x 0,75)
Hàm lượng % HQ và % DTBHQ đã được xác định ở trên.
QUY ĐỊNH ĐÁNH GIÁ VỆ SINH AN TOÀN BUTYL HYDROXYANISOL
1. Tên khác, ký hiệu BHA; Chỉ số INS: 320; Chỉ số EEC: E320; Butylated hydroxyanisol;
2. Định danh
Tên hóa học 2 (3) - Tertiary-butyl-4-hydroxyanisol; hỗn hợp của 3 và 2- tertiary-butyl-4-hydroxyanisol
Mã số C.A.S. 25013-16-5
Công thức hóa học C11H16O2
| Công thức phân tử |
|
Khối lượng phân tử 180,25
3. Mô tả ngoại quan Tinh thể trắng, vàng nhạt hoặc là dạng sáp rắn, có mùi đặc trưng nhẹ
4. Chức năng Chất chống oxy hóa
5. Yêu cầu về vệ sinh an toàn
5.1. Định tính
Tính tan Không tan trong nước, tan tốt trong etanol và propan-1,2-diol
Phản ứng màu Đạt yêu cầu (mô tả trong phương pháp kiểm thử)
5.2. Độ tinh khiết
Tro sulfat Không lớn hơn 0,05%
Asen Không lớn hơn 3mg/kg
Tổng các kim loại nặng Không lớn hơn 10mg/kg
Tạp phenol Không lớn hơn 0,5%
5.3. Định lượng C11H16O2 Không nhỏ hơn 98,5% C11H16O2 và không nhỏ hơn 85% 3-Tert-butyl-4-hydroxyanisol
6. Phương pháp thử
6.1. Thử định tính
Thử phản ứng màu
Hóa chất:
Dung dịch 2,6 - dicloroquinonclorimit 0,01%: Cân 0,5g 2,6 - dicloroquinon-clorimit pha trong cồn tuyệt đối, định mức tới 50ml bằng cồn tuyệt đối. Lấy 1ml dung dịch này pha loãng bằng cồn tuyệt đối và định mức tới 100ml
Tiến hành
Hòa tan 1 mẫu trong 100ml etanol 72%. (Dung dịch A)
Lấy 1ml dung dịch A cho vào bình định mức 100ml, định mức tới vạch bằng etanol 72% (dung dịch thử)
Lấy 5ml dung dịch thử, thêm 2ml dung dịch natri borat TT.222 và 1ml dung dịch 2,6 - dicloroquinonclorimid 0,01%, trộn đều hỗn hợp. Dung dịch phải xuất hiện mầu xanh.
6.2. Thử độ tinh khiết
Xác định tro sulfat
Xem hướng dẫn chung các phương pháp thử (P.VC.11)
Chú ý: Lấy 5g mẫu thử
Xác định asen
Xem hướng dẫn chung các phương pháp thử (P.VC.08)
Xác định tổng các kim loại nặng
Xem hướng dẫn chung các phương pháp thử (P.VC.10-Phương pháp 2)
Xác định tạp phenol
Xác định bằng sắc ký lớp mỏng: dùng bản mỏng silicagel G.
Thuốc thử:
- Sắt (3) clorid 2%: Cân 2g FeCl3 hoà tan và định mức tới 100ml bằng nước cất
- Kali ferocyanid 1%: Cân 1g kali ferocyanid hoà tan và định mức tới 100ml bằng nước cất.
- Dung dịch thuốc hiện màu: Trộn dung dịch sắt clorid 2% với dung dịch kali ferocyanid 1% với tỷ lệ 1/1
Tiến hành:
- Dung dịch 1: Hòa tan 0,25g mẫu thử cần xác định trong 10ml ete
- Dung dịch 2: Pha loãng 1ml dung dịch 1 tới 10ml với ete và tiếp theo pha loãng 1ml dung dịch này tới 20ml với ete. Sử dụng dung dịch pha loãng cuối cùng là dung dịch 2.
Lần lượt chấm 2μl dung dịch 1 và dung dịch 2 trên các bản sắc ký riêng biệt. Đặt bản mỏng vào trong bình khai triển sắc ký có bão hòa dung môi cloroform. Để dung môi khai triển cách vạch xuất phát 15cm. Lấy bản mỏng ra khỏi bình sắc ký, để ráo bản mỏng, rồi phun sương thuốc thử hiện màu. Trên bản mỏng xuất hiện các đốm màu xanh, các đốm này sẽ hiện rõ hơn bằng cách phun sương acid hydrocloric 2N lên bản mỏng.
Bất kỳ vết mầu xanh xuất hiện trên sắc ký đồ của dung dịch 1 (ngoài vết chính và vết có Rf = 0,35) phải nhạt màu hơn so với vết chính xuất hiện trong sắc ký đồ của dung dịch 2.
6.3. Định lượng C11H16O2
Phương pháp sắc ký khí
Dung dịch nội chuẩn: (có thể tuỳ chọn Diphenylamin hoặc 4-tert-butylphenol)
Cân chính xác 500mg chất nội chuẩn hòa tan và định mức tới 250ml bằng aceton
Dung dịch chuẩn:
Cân chính xác 90mg 3-butyl hydroxyanisol và 10mg 2-butyl hydroxyanisol, hòa tan và định mức đến 100ml trong dung dịch nội chuẩn. Pha các nồng độ để xây dựng đường chuẩn theo yêu cầu.
Tiến hành:
• Cân chính xác 10mg mẫu thử BHA, hòa tan và định mức đến 50ml bằng trong dung dịch nội chuẩn.
• Sử dụng thiết bị sắc ký khí với detetor ion hóa ngọn lửa. Điều kiện làm việc như sau:
- Điều kiện để chất nội chuẩn rửa giải ra sau 3-tert-butyl-4-hydroxyanisol:
Cột: Cột thủy tinh kích thước 2mm x 1,5m
Chất nhồi: 10%XE-60 cỡ hạt 100 - 200mesh
Nhiệt độ cột: 1550C
Nhiệt độ detector: 2500C
Nhiệt độ buồng bơm mẫu: 2500C
Khí mang: N2, tốc độ dòng 30ml/phút
- Điều kiện để chất nội chuẩn rửa giải ra trước 3-ter-butyl-4-hydroxyanisol:
Cột: Cột thuỷ tinh kích thước 3mm x 2m
Chất nhồi: 5% Versamit-900 cỡ hạt 80/100 mesh cromosorb W-AW-DMCS
Nhiệt độ cột: 1700C
Nhiệt độ buồng bơm: 2250C
Nhiệt độ detector: 2500C
Khí mang: N2, tốc độ dòng 30ml/phút
Lần lượt bơm các dung dịch chuẩn theo nồng độ tăng dần. Dựng đường chuẩn biểu thị tương quan giữa chiều cao pic sắc ký với nồng độ dung dịch chuẩn.
Bơm dung dịch mẫu. Dựa vào đường chuẩn xác định Nồng độ 2- và 3-butyl hydroxyanisol
• Chuẩn bị mẫu thử: cân chính xác khoảng100mg mẫu hòa tan và pha loãng tới 10ml với dung dịch chuẩn nội.
• Hệ thống sắc ký: Hệ thống sắc ký khí bao gồm detector ion hóa ngọn lửa và cột bằng thép không gỉ kích thước 1,8m x 2mm, được nhồi chất mang tẩm 10% pha tĩnh lỏng, cột được luyện trong chế độ đẳng nhiệt ở nhiệt độ trong khoảng 175 đến 1850C, khí mang sử dụng là heli. Bơm dung dịch chuẩn vào máy, ghi lại pic để đảm bảo chắc chắn rằng độ lệch chuẩn không vượt quá 2% với đồng phân 3-tert-butyl-4-hydroxyanisol và không vượt quá 6% với đồng phân 2-tert-butyl-4-hydroxyanisol. Hệ số phân giải giữa các đồng phân không nhỏ hơn 1,3 và hệ số roãng pic không được vượt quá 2%.
• Pha tĩnh lỏng: 25% 2-cyanoethyl và 75% methyl-polysiloxan
• Chất mang: Đất silic dùng cho sắc ký được nung chảy bằng cách trộn hỗn hợp diatomit với Na2CO3 nung chảy và nung ở 9000C. Đất silic được rửa bằng acid, sau đó được rửa bằng nước cho đến khi trung tính nhưng không được rửa bằng bazơ. Đất silic được silan hóa bằng cách xử lý với hóa chất như dimetyldiclorosil để che phủ cho nhóm hoạt động bề mặt silanol.
• Tiến hành: bơm lần lượt (khoảng 5μl) dung dịch chuẩn và dung dịch chuẩn bị thử nghiệm vào máy sắc ký khí, ghi lại sắc ký đồ. Đo diện tích peak của mỗi đồng phân và dung dịch chuẩn nội trong mỗi sắc ký đồ.
Tính kết quả:
Khối lượng (mg) của mỗi đồng phân trong mẫu thử theo công thức:
Lượng đồng phân trong mẫu thử = (mg)
Trong đó:
Cs: nồng độ của đồng phân trong dung dịch chuẩn
Rs: tỷ số diện tích của mỗi đồng phân so với chất nội chuẩn trong sắc ký đồ với dung dịch chuẩn.
R0: tỷ số diện tích của mỗi đồng phân của dung dịch nội chuẩn trong sắc ký đồ với dung dịch thử nghiệm.
QUY ĐỊNH ĐÁNH GIÁ VỆ SINH AN TOÀN BUTYL HYDROXYTOLUEN
1. Tên khác, ký hiệu BHT; Chỉ số INS: 321; Chỉ số EEC: E 321; Butylated hydroxytoluen;
2. Định danh
Tên hóa học 2,6-ditertiary-butyl-p-cresol, 4-metyl-2,6-ditertiary-butyl-phenol
Mã số C.A.S 128-37-0
Công thức hóa học C15H24O
| Công thức phân tử |
|
Khối lượng phân tử 220,36
3. Mô tả ngoại quan Dạng tinh thể hoặc vảy rắn, màu trắng, không mùi hoặc có mùi thơm nhẹ đặc trưng
4. Chức năng Chống oxy hóa
5. Yêu cầu về vệ sinh an toàn
5.1. Định tính
Tính tan Không tan trong nước và propan-1,2-diol. Tan tốt trong etanol
Khoảng nhiệt độ nóng chảy 690 - 720C
Bước sóng hấp thụ cực đại Đạt yêu cầu (mô tả trong phần phương pháp thử)
Phản ứng mầu Đạt yêu cầu (mô tả trong phần phương pháp thử)
5.2. Độ tinh khiết
Nhiệt độ đông đặc Không nhỏ hơn 69,20
Tro sulfat Không lớn hơn 0,005%
Asen Không lớn hơn 3mg/kg
Tổng các kim loại nặng Không lớn hơn 10mg/kg
Tạp phenol Không lớn hơn 0,5%
5.3. Định lượng C15H24O Không nhỏ hơn 99,0%
6. Phương pháp thử
6.1. Thử định tính
Xác định khoảng nhiệt độ nóng chảy
Xem hướng dẫn chung các phương pháp thử
Xác định bước sóng hấp thụ cực đại
Pha dịch mẫu thử trong etanol khan nồng độ 1/100.000 (dịch thử)
Đo độ hấp thụ quang của dịch thử trên toàn dải bước sóng từ 230nm đến 320nm (cuvet đo quang có chiều dày 2cm). Phổ hấp thụ quang của dịch thử chỉ đạt 1 cực đại hấp thụ duy nhất tại 278nm.
Phản ứng màu
Hóa chất:
- Dung dịch NaNO2 0,3%: Cân 0,3g NaNO2 hoà tan và định mức tới 100ml bằng nước cất.
- Thuốc thử dianisidin dihydroclorid: Cân 200mg 3,3 dimetoxy - benzidin dihydroclorid hòa tan trong hỗn hợp 40ml metanol và 60ml HCl 1N
Tiến hành:
Hoà tan 10ml dung dịch mẫu thử 0,001% trong metanol thêm 10ml nước cất, 2ml dung dịch NaNO2 0,3% và 5ml thuốc thử dianisidin dihydroclorid. Trong 3 phút dung dịch sẽ xuất hiện sẽ xuất hiện màu đỏ cam. Thêm 5ml cloroform và lắc đều. Lớp cloroform xuất hiện mầu tím hoặc đỏ tươi, mầu này bị phai dần khi đưa ra ánh sáng.
6.2. Thử độ tinh khiết
Xác định nhiệt độ đông đặc
Xem hướng dẫn chung các phương pháp thử (P.VL.02)
Xác định tro sulfat
Xem hướng dẫn chung các phương pháp thử (P.VC.11)
Chú ý: Lấy 20g mẫu thử
Xác định asen
Xem hướng dẫn chung các phương pháp thử (P.VC.08)
Xác định tổng các kim loại nặng
Xem hướng dẫn chung các phương pháp thử (P.VC.10-Phương pháp 2)
Chú ý: Lấy 2g mẫu thử
Xác định tạp phenol
Xác định bằng sắc ký lớp mỏng: dùng bản mỏng silicagel G.
Chuẩn bị thuốc thử:
- Sắt (3) clorid 2%: cân 2g FeCl3 hòa tan và định mức tới 100ml bằng nước cất.
- Kali ferocyanid 1%: Cân 1g Kali ferocyanid hòa tan và định mức tới 100ml bằng nước cất.
- Dung dịch thuốc hiện màu: Trộn dung dịch sắt clorid 2% với dung dịch kali ferocyanid 1% với tỷ lệ 1/1
Tiến hành:
- Dung dịch 1: Hoà tan 0,25g mẫu thử cần xác định trong 10ml ete
- Dung dịch 2: pha loãng 1ml dung dịch 1 tới 10ml với ete và tiếp theo pha loãng 1ml dung dịch này tới 20ml với ete. Sử dụng dung dịch pha loãng cuối cùng là dung dịch 2.
- Lần lượt chấm 2μl dung dịch 1 và dung dịch 2 trên các bản sắc ký riêng biệt. Đặt bản mỏng vào trong bình khai triển sắc ký đã bão hòa dung môi cloroform. Để dung môi khai triển cách vạch xuất phát 15cm. Lấy bản mỏng ra khỏi bình sắc ký, để ráo bản mỏng, rồi phun sương thuốc thử hiện màu. Trên bản mỏng xuất hiện các đốm màu xanh, các đốm này sẽ hiện rõ hơn bằng cách phun sương acid hydrocloric 2N lên bản mỏng.
Bất kỳ vết mầu xanh xuất hiện trên Sắc ký đồ của dung dịch 1 (ngoài vết chính và vết có Rf = 0,35) phải nhạt màu hơn so với vết chính xuất hiện trong sắc ký đồ của dung dịch 2.
6.3. Định lượng C15H24O
Phương pháp sắc ký khí
Dung dịch nội chuẩn:
(chất nội chuẩn tuỳ chọn diphenylamin hoặc 4-tert butylphenol)
Cân chính xác 50mg butyl hydroxytoluen hoà tan trong aceton và dùng aceton định mức cho đủ 250ml
Dung dịch chuẩn:
Cân chính xác 100mg butyl hydroxytoluen hòa tan trong aceton và dùng aceton định mức cho đủ 50ml
Tiến hành thử:
Cân chính xác 10mg mẫu thử, hoà tan vào dung dịch nội chuẩn và định mức đến 50ml. Bơm dung dịch này vào sắc ký khí với detector ion hóa ngọn lửa hydro. Cột thuỷ tinh kích thước 1,5m x 3mm 10% XE - 60 trên 100 - 2000 mesh aeropak 30, hoặc thiết bị tương đương, cột được luyện với chế độ đẳng nhiệt tại 1550C. Nhiệt độ của buồng bơm mẫu và của detector tương ứng là 2250C và 2500C. Khí mang là nitơ với tốc độ dòng 30ml/phút.
Dựng đồ thị biểu diễn tỷ số chiều cao pic butyl hydroxytoluen/chiều cao pic của dung dịch nội chuẩn tại các nồng độ xác định khác nhau. Nồng độ của butyl hydroxytoluen được xác định bằng nội suy từ đồ thị chuẩn.
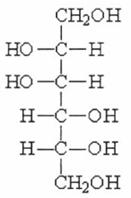
QUY ĐỊNH ĐÁNH GIÁ VỆ SINH AN TOÀN MANNITOL
1. Tên khác, ký hiệu d-mannitol, mannit; Chỉ số INS: 421; Chỉ số EEC: E421
2. Định danh
Tên hóa học D-mannitol
Mã số C.A.S. 69-65-8
Công thức hóa học C6H14O6
| Công thức phân tử |
|
Khối lượng phân tử 182,17
3. Mô tả ngoại quan Dạng bột tinh thể, không mùi, màu trắng, vị ngọt
4. Chức năng Tạo ngọt, làm ẩm, ổn định, chất độn
5. Yêu cầu về vệ sinh an toàn
5.1. Định tính
Tính tan Tan trong nước, rất ít tan trong etanol, hầu như không tan ete
Khoảng nhiệt độ nóng chảy Từ 1650C đến 1690C
Sắc ký bản mỏng Vết chính trong sắc ký đồ bản mỏng của mẫu thử phải tương ứng về Rf, màu sắc và kích cỡ với vết chính trong sắc ký đồ bản mỏng của chất chuẩn.
5.2. Độ tinh khiết
Giảm khối lượng khi làm khô Không lớn hơn 0,3% (1050C; 4 giờ)
Góc quay cực riêng α20D = +230 đến +250 (trong dung dịch Borat)
Độ pH 5 - 8 (dung dịch 10% m/v)
Tro sulfat Không lớn hơn 0,1%
Clorid Không lớn hơn 70mg/kg
Sulfat Không lớn hơn 100mg/kg
Niken Không lớn hơn 2mg/kg
Chì Không lớn hơn 1mg/kg
Tổng các kim loại nặng Không lớn hơn 10mg/kg
Đường thử Không lớn hơn 0,3%
Đường tổng số Không lớn hơn 1,0%
5.3. Định lượng C6H14O6 Từ 96,0% đến 102% D-mannitol (tính theo chế phẩm khan)
6. Phương pháp thử
6.1. Thử định tính
Xác định khoảng nhiệt độ nóng chảy
Xem hướng dẫn chung các phương pháp thử (P.VL.01)
Kiểm tra sắc ký bản mỏng
Bản mỏng:
Bản mỏng tráng sẵn pha tĩnh là silicagen
Pha động
Propanol: etyl acetat: nước (70: 20: 10 - v:v:v)
Dung dịch chuẩn:
Hòa tan 50mg Mannitol trong 20ml nước cất.
Dung dịch thuốc thử:
- Thuốc thử Acid 4-aminobenzoic: cân 1g acid 4-aminobenzoic hòa tan trong dung môi gồm: 18ml acid acetic, 20ml nước và 1ml acid phosphoric. Sau đó trộn dung dịch này với aceton tỷ lệ 2:3 (v:v).
- Thuốc thử Natri periodat: pha dung dịch Natri periodat trong nước cất nồng độ 0,2% (m/v)
Tiến hành:
Hoà tan 50mg mẫu thử trong 20ml nước cất. Chấm khoảng 2μl chất chuẩn và dung dịch mẫu thử vào vạch xuất phát của bản mỏng. Khai triển sắc ký lên đến độ cao cách vạch xuất phát khoảng 17cm. Để bản mỏng khô trong không khí và phun thuốc thử acid 4-aminobenzoic lên bản mỏng. Sấy bản mỏng tại 1000C, trong 15 phút. Phun sương thuốc thử natri periodat. Sấy bản mỏng tại 1000C, trong 15 phút.
Đánh giá kết quả:
Vết chính trong sắc ký đồ của mẫu phải tương đương về vị trí, màu và kích thước so với vết chính trong sắc ký đồ của chuẩn.
6.2. Thử độ tinh khiết
Xác định giảm khối lượng khi làm khô
Xem hướng dẫn chung các phương pháp thử (P.VC.01)
Chú ý: Mẫu được sấy tại 1050C trong 4 giờ
Xác định góc quay cực riêng
Cân chính xác 2g mẫu và 2,6g dinatri tetraborat, hoà tan trong 20ml nước nóng khoảng 300C, lắc đều trong 15 - 30 phút (không đun nóng tiếp) thu được dung dịch trong. Pha loãng tới 25ml bằng nước cất.
Xem hướng dẫn chung các phương pháp thử (P.VL.03)
Xác định độ pH
Pha dung dịch mẫu 10% (m/v), thêm 0,5ml KCl bão hoà. Tiến hành đo pH.
Xác định tro sulfat
Xem hướng dẫn chung các phương pháp thử (P.VC.11)
Chú ý: lấy 2g mẫu thử
Xác định clorid
Xem hướng dẫn chung các phương pháp thử (P.VC.04)
Chú ý: Lấy 10g mẫu thử. Dùng 2ml dung dịch HCl 0,01N làm dung dịch chuẩn.
Xác định sulfat
Xem hướng dẫn chung các phương pháp thử (P.VC.05)
Chú ý: Lấy 10g mẫu thử. Dùng 2ml dung dịch H2SO4 0,01N làm dung dịch chuẩn
Xác định niken
Phương pháp quang phổ hấp thụ nguyên tử ngọn lửa
Hóa chất:
Dung dịch amoni pyrolidin-dithiocacbamat
Thiết bị:
Máy quang phổ hấp thụ nguyên tử: hoạt động theo các điều kiện sau:
- Vạch phổ đo: 232,0nm.
- Cường độ đèn: 70% Imax
- Khe đo: 0,5nm
- Tốc độ không khí nén: 4,5ml/phút
- Tốc độ axetylen: 1,2ml/phút
- Các điều kiện khác tuỳ theo yêu cầu riêng cho mỗi loại thiết bị
Dung dịch thử
Hoà tan 20g mẫu thử vào hỗn hợp của dung dịch acid acetic loãng TT.002 và nước cất (1:1) và định mức tới 100ml bằng hỗn hợp đó. Thêm 2ml dung dịch ammoni pyrolidin dithiocacbamat 1% và 10ml metyl isobutyl keton. Lắc, để yên cho tách lớp. Sử dụng lớp metyl isobutyl keton.
Dung dịch chuẩn:
Thêm 0,5ml; 1ml; 1,5ml dung dịch chuẩn nickel nồng độ 10mg/kg tương ứng vào 3 dung dịch chuẩn được chuẩn bị giống như dung dịch thử.
Tiến hành:
Sử dụng metyl isobutyl keton làm mẫu trắng, hiệu chỉnh máy về 0. Tiến hành đo mẫu tại điều kiện mô tả như trên.
Xác định chì
Xem hướng dẫn chung các phương pháp thử (P.VC.09).
Xác định tổng các kim loại nặng
Xem hướng dẫn chung các phương pháp thử (P.VC.10 - Phương pháp 1).
Chú ý: Dung dịch thử: Hoà tan 2g mẫu trong 25ml nước cất.
Xác định đường thử
Xem hướng dẫn chung các phương pháp thử (P.HC.03)
Đánh giá kết quả:
Lượng Cu2O thu được không được vượt quá 50mg
Xác định đường tổng số
Cho 2,1g mẫu thử vào bình 250ml, thêm 40ml HCl 0,1N. Nối bình với sinh hàn hồi lưu, cất hồi lưu trong 4 giờ. Chuyển dung dịch vào cốc có mỏ 400ml, rửa bình bằng 10ml nước cất. Trung hòa dung dịch bằng NaOH 6N.
Tiến hành tiếp như chi tiết đã mô tả ở phần xác định hàm lượng đường thử.
Đánh giá kết quả:
Lượng Cu2O thu được không vượt quá 50mg.
6.3. Định lượng mannitol
Phương pháp sắc ký lỏng phân giải cao (HPLC)
Thiết bị:
• Máy Sắc ký lỏng (HPLC)
- Detector: detector khúc xạ đo ở nhiệt độ không đổi
- Cột sắc ký: AMINEX HPX 87C, L = 30cm, ID = 9mm
- Dung môi và nước cất 2 lần đã deion hóa (đã được lọc qua màng lọc cỡ 0,45μm)
• Điều kiện sắc ký:
- Nhiệt độ cột: 85 ± 0,50C
- Tốc độ dòng: 0,5ml/phút
Dung dịch chuẩn:
Hòa tan khối lượng chính xác của chuẩn mannitol trong nước cất để thu được một dung dịch có nồng độ 10mg/ml
Dung dịch mẫu thử:
Cân chính xác 1g mẫu thử, cho vào bình định mức 50ml, hoà tan và định mức đến vạch bằng nước cất.
Tiến hành đo:
Bơm lần lượt 20μl dung dịch chuẩn và dung dịch mẫu thử đã chuẩn bị vào máy sắc ký, ghi lại sắc ký đồ và đo độ đáp ứng của pik mannitol
Tính kết quả:
Hàm lượng mannitol trong phần mẫu thử đã lấy ra theo công thức:
Trong đó:
ms: khối lượng mannitol trong mẫu thử (mg).
C: Nồng độ mannitol (mg/ml) trong dung dịch chuẩn
RU: diện tích pik thu được trên sắc ký đồ của dung dịch mẫu thử
Rs: diện tích pik thu được trên sắc ký đồ của dung dịch chuẩn.
QUY ĐỊNH ĐÁNH GIÁ VỆ SINH AN TOÀN ASPARTAM
1. Tên khác, ký hiệu Aspartyl phenylalanin metyl este, APM; Chỉ số INS: 951
2. Định danh
Tên hóa học 3-Amino-N-(α-cacbomethoxy-phenetyl)-acid succinamic, N-L-α-aspartyl-L-phenylalanin-1-metyl este
Mã số C.A.S 22389-47-0
Công thức hóa học C14H18N2O5
| Công thức phân tử |
|
Khối lượng phân tử 294,31
3. Mô tả ngoại quan Dạng bột tinh thể màu trắng, không mùi, có vị ngọt mạnh
4. Chức năng Chất tạo ngọt
5. Yêu cầu về vệ sinh an toàn
5.1. Định tính
Tính tan ít tan trong nước và etanol
Định tính nhóm amin Đạt (mô tả trong phương pháp thử)
Định tính nhóm este Đạt (mô tả trong phương pháp thử)
5.2. Độ tinh khiết
Giảm khối lượng khi làm khô Không lớn hơn 4,5% (1050C; 4 giờ)
Độ pH 4,5 đến 6,0
Góc quay cực riêng [α]D20: +14,50 đến 16,50 (tính theo chế phẩm khan)
Độ truyền qua Đạt (mô tả trong phương pháp thử)
Tro sulfat Không lớn hơn 0,2%
Asen Không lớn hơn 3mg/kg
Tổng các kim loại nặng Không lớn hơn 10mg/kg
| (5-benzyl-3-6-dioxo-2-piperazin acetic) | Không lớn hơn 1,5% |
Đồng phân quang học khác Tổng hàm lượng L-α-aspartyl-D-phenylalanin metyl este và D-α-aspartyl-L-phenylalanin metyl este không lớn hơn 1mg/kg.
5.3. Định lượng C14H18N2O5 Không nhỏ hơn 98% C14H18N2O5 (tính theo chế phẩm khan)
6. Phương pháp thử
6.1. Thử định tính
Thử định tính nhóm amino
Thuốc thử:
Hoà tan 2g ninhydrin trong 75ml dimetylsunfoxid, cho vào bình định mức 100ml, thêm 62mg hydrindantin, thêm dung dịch đệm lithi acetat 4M (pH = 9) tới 100ml lắc kỹ và lọc.
Tiến hành:
Cho 10g mẫu thử vào ống nghiệm, thêm 2ml dung dịch thuốc thử và đun nóng. Dung dịch phải chuyển thành màu tím đậm.
Thử định tính nhóm este
Hoà tan khoảng 20mg mẫu trong 1ml metanol thêm 0,5ml dung dịch metanol bão hòa hydroxylamin clohydric, trộn hỗn hợp và tiếp tục thêm 0,3ml KOH 5N. Đun sôi hỗn hợp, để nguội, điều chỉnh tới pH =1-1,5 bằng dung dịch acid hydrocloric TT.266, thêm 0,1ml dung dịch sắt (III) clorid TT.112. Dung dịch phải tạo thành màu đỏ tía.
6.2. Thử độ tinh khiết
Xác định giảm khối lượng khi làm khô
Xem hướng dẫn chung các phương pháp thử (P.VC.01)
Ghi chú: Mẫu được sấy tại 1050C trong 4 giờ
Xác định độ pH
Dịch thử pH: dung dịch mẫu thử nồng độ 1/125
Xác định góc quay cực riêng
Xem hướng dẫn chung các phương pháp thử (P.VL.03)
Ghi chú: Pha dung dịch mẫu thử 4% trong acid formic 15N; tiến hành đo trong 30 phút sau khi pha dung dịch mẫu thử.
Xác định độ truyền qua
Chuẩn bị dung dịch thử:
Cân 1g mẫu thử, hòa tan trong HCl 2N, cho vào bình định mức 100ml, định mức đến 100ml bằng HCl 2N.
Đo độ truyền qua của dung dịch thử trên máy. Dung dịch so sánh là HCl 2N
Đánh giá kết quả:
Hệ số truyền qua không nhỏ hơn 0,95 tương ứng với độ hấp thụ không lớn hơn 0,022
Xác định tro sulfat
Xem hướng dẫn chung các phương pháp thử (P.VC.11)
Chú ý: Lấy 1g mẫu thử
Xác định asen
Xem hướng dẫn chung các phương pháp thử (P.VC.08)
Xác định tổng kim loại nặng
Xem hướng dẫn chung các phương pháp thử (P.VC.10-Phương pháp 2)
Chú ý: Lấy 2g mẫu thử
Xác định 5-benzyl-3-6-dioxo-2-piperazineacetic
Thiết bị:
• Sử dụng thiết bị sắc ký khí, detector ion hóa ngọn lửa và được thiết kế cột thủy tinh với buồng bơm thẳng (Micro-Tex 220 hoặc tương đương). Cột thuỷ tinh kích thước 1,83m x 4mm, pha tĩnh nhồi diatomit trơ (3% OV-1 trên nền pha tĩnh supelcoport cỡ hạt 80/100mesh). Thiết bị được cần được chạy ổn định tại 2500C qua đêm. Để ngăn ngừa sự cháy của oxid silic, làm sạch detector với aceton thường xuyên.
• Điều kiện làm việc: Thông số hoạt động có thể biến đổi, tuỳ thuộc vào hệ thống thiết bị sử dụng, nhưng thông thường sử dụng những điều kiện sau:
- Nhiệt độ cột: 2000C
- Nhiệt độ buồng bơm: 2000C
- Nhiệt độ detector: 2750C
- Khí mang: heli với tốc độ dòng 75ml/phút
- Dòng hydro và không khí nén cho detector, đặt tốc độ sao cho độ nhạy detector đạt lớn nhất.
Thuốc thử silan hóa:
Chỉ chuẩn bị trước khi dùng: Pha loãng 3 phần, theo thể tích, của N,O-bis-(trimetylsilyl) axetamit với 2 phần dimetylformamit.
Dung dịch chuẩn:
Cân chính xác 25mg acid 5-benzyl-3,6-dioxo-2-piperazon acetic chuẩn chuyển vào bình định mức 50ml, pha loãng đến vạch mức bằng metanol, trộn hỗn hợp. Hút 10ml dung dịch này vào bình định mức 100ml, pha loãng đến vạch với metanol, trộn hỗn hợp. Lấy 3ml dung dịch thứ hai cho vào ống đựng mẫu, đóng nắp teflon rồi cô đến khô trong nồi cách thủy. Thêm 1ml thuốc thử silan hóa vào phần cặn, đậy nắp ống đựng mẫu, lắc rồi đun nóng trong lò ở 800C trong 30 phút. Nhấc ống đựng mẫu ra khỏi lò, lắc 15 giây và làm nguội tới nhiệt độ phòng.
Dung dịch mẫu thử:
Cân chính xác khoảng 10mg mẫu Aspartam chuyển vào trong ống đựng mẫu với nắp kín teflon, thêm 1ml thuốc thử silla hóa, đậy nắp, lắc và làm nóng trong lò với 800C trong 30 phút. Chuyển ống đựng mẫu ra khỏi lò và làm nguội đến nhiệt độ phòng.
Tiến hành:
Bơm 3μl dung dịch chuẩn vào máy sắc ký khí, ghi sắc ký đồ, đo chiều cao pic của acid 5-benzyl-3,6-dioxo-2-piperazine acetic (hs). Ở cùng điều kiện, thời gian rửa giải khoảng 7-9 phút. Bơm 3μl của dung dịch mẫu thử, ghi sắc đồ, đo chiều cao pic của acid 5-benzyl-3,6-dioxo-2-piperazin acetic có trong mẫu thử (h).
Tính kết quả:
Tính % của acid 5-benzyl-3,6-dioxo-2-piperazin acetic trong mẫu bằng công thức sau:
% acid 5-benzyl-3,6-dioxo-2-piperazin acetic = x 100
Trong đó:
ms: khối lượng của chuẩn (mg)
m: khối lượng của mẫu Aspartam (mg)
Xác định đồng phân quang học khác
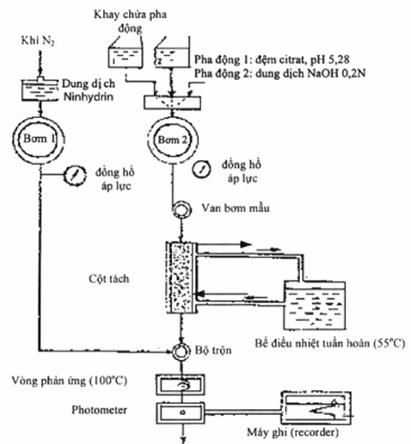
Thiết bị:
Sử dụng thiết bị phân tích acid amin (VD: Hitachi KLA-5 hoặc tương đương). Cột tách: 550mm, đường kính 9mm; pha tĩnh là nhựa trao đổi cation mạnh (khoảng 50g) (Nhựa trao đổi ion của Hitachi No.2613 hoặc tương đương). Cột phản ứng dài 29m, đường kính 0,5mm, detector đo quang có thể đo được ở bước sóng 570nm.
Điều kiện hoạt động:
Các thông số hoạt động có thể thay đổi tuỳ thuộc vào hệ thống thiết bị sử dụng
- Nhiệt độ cột tách: 550C
- Nhiệt độ buồng phản ứng dẫn xuất sau cột: 1000C
- Dung môi rửa giải: dung dịch đệm citrat có pH = 5,28
- Áp suất rửa giải: 8 - 10kg/cm2
- Tốc độ dòng: 60ml/phút
- Áp suất của dung dịch ninhydrin: 2 - 5 kg/cm2
- Tốc độ dòng của dung dịch ninhydrin: 30ml/giờ
- Detector: đo ở bước sóng 570nm
Sơ đồ thiết bị như sau:
Hóa chất và thuốc thử:
- Dung dịch đệm citrat pH = 5,28: hòa tan 34,3g natri citrat (C6H5Na3O2.2H2O) trong khoảng 400ml nước cất, thêm 7,5ml dung dịch acid hydrocloric TT.266 và 5ml benzyl alcohol, và thêm nước cất đủ đến mức 1000ml.
- Dung dịch đệm citrat pH = 2,2; Hoà tan 1,4g natri citrat (C6H5Na3O2.2H2O), 13g acid citric (C6H8O2.H2O) và 10,9g NaCl trong khoảng 400ml nước cất và định mức vừa đủ tới 1000ml.
- Dung dịch ninhydrin: Rót 140ml nước cất vào bình tam giác 500ml, thêm 82,0g natri acetat (C2H3NaO2), khuấy cho hòa tan hoàn toàn, thêm 25ml acid acetic băng và pha loãng tới 250ml với nước cất. Điều chỉnh dung dịch tới pH = 5,51 ± 0,03 bằng acid acetic băng hoặc dung dịch natri acetat TT.218 (dung dịch đệm acetat). Rót 750ml dung dịch metyl-cellosolven vào trong lọ thuỷ tinh nấu 1000ml và thêm 250ml dung dịch đệm acetat. Cho khí nitơ đi qua dung dịch trong khi vẫn lắc hỗn hợp. Hòa tan 20g ninhydrin và 0,38g thiếc clorid (SnCl2.H2O) vào trong dung dịch. Để yên dung dịch 24 giờ trước khi dùng.
Dung dịch chuẩn:
Chuyển 0,25mg L-α-aspartyl-D-phenylalanin metyl este chuẩn vào trong bình định mức 100ml. Hòa tan và định mức với nước cất (dung dịch A). Chuyển 250mg của L-α-aspartyl-D-phenylalanin metyl este chuẩn vào trong bình định mức 100ml khác, hòa tan trong dung dịch đệm pH = 2,2. Thêm 10ml dung dịch A và pha loãng tới mức với dung dịch đệm pH = 2,2. Giữ dung dịch này ở nhiệt độ dưới 50C.
Dung dịch mẫu thử:
Chuyển 250mg mẫu thử Aspartam cần xác định vào bình định mức 100ml, hòa tan và pha loãng tới mức với dung dịch đệm pH = 2,2.
Tiến hành:
Luyện cột bằng dung dịch NaOH 0,2N và tiếp theo với dung dịch đệm citrat pH = 5,28. Sau đó dẫn dung dịch ninhydrin vào hệ thống. Bơm 500μl dung dịch chuẩn vừa chuẩn bị ở trên vào máy phân tích acid amin, ghi sắc đồ. Trong cùng một điều kiện, thời gian lưu tương ứng của L-α-aspartyl-D-phenylalanin metyl este là 100 phút, của L-α-aspartyl-L-phenylalanin metyl este là 115 phút.
Bơm 500μl dung dịch mẫu thử vừa chuẩn bị và ghi sắc đồ. So sánh sắc đồ này với sắc đồ dung dịch chuẩn và xác định thành phần bằng cách so sánh thời gian lưu. Không được có pic phù hợp với L-α-aspartyl-D-phenylalanine metyl este. (Phương pháp này cũng áp dụng cho D-α-aspartyl-L-phenylalanin metyl este). Giới hạn phát hiện của tổng L-α-aspartyl-D-phenylalanin metyl este và D-α-aspartyl-L-phenylalanin metyl este là 1μg/ml.
6.3. Định lượng Aspartam
Cân chính xác khoảng 150mg mẫu thử Aspartam cần xác định, làm khô ở 1500C trong 4 giờ, hoà tan trong 35ml dimetylformamit, thêm 5 giọt thuốc thử thymol xanh TT.251 và chuẩn độ bằng microburet với dung dịch liti methoxid 0,1N tới màu xanh đậm. Thực hiện đối với màu trắng để xác định hệ số điều chỉnh cần thiết.
1ml dung dịch liti methoxid 0,1N tương đương với 29,43mg C14H18N2O5
Chú ý: Để tránh dung dịch không hấp thụ CO2 và hút ẩm, khi hòa tan mẫu và trong quá trình chuẩn độ phải bọc bình chuẩn độ bằng giấy nhôm.
QUY ĐỊNH ĐÁNH GIÁ VỆ SINH AN TOÀN ISOMALT
1. Tên khác, ký hiệu Isomanitol, hydrogenated isomaltulose, hydrogenerated palatinose; Chỉ số INS: 953
2. Định danh
Tên hóa học Isomalt là hỗn hợp của mono- và di-saccarid hydro hóa, thành phần chính là những disaccarid sau: 6-O-α-D-Glucopyranosyl-D-sorbitol (1,6-GPS) và 1-O-α-D-Glucopyranosyl-D-mannitol dihydrat (1,1-GPM)
Mã số C.A.S. 64519-82-0
Công thức hóa học 6-O-α-D-Glucopyranosyl-D-sorbitol: C12H24O11; 1-O-α-D-Glucopyranosyl-D-mannitol dihydrat: C12H24O11.2H2O
Công thức phân tử
|
|
|
| 6-O-alpha-D-Glucopyranosyl-D-sorbitol (1,6-GPS) | 1-O-alpha-D-Glucopyranosyl-D-mannitol (1,1-GPM) |

Khối lượng phân tử 1,6-GPS: 344.32; 1,1-GPM: 380.32
3. Mô tả ngoại quan Dạng tinh thể, không mùi, màu trắng, vị ngọt
4. Chức năng Tạo ngọt, chất độn, chất chống đông vón, chất làm bóng
5. Yêu cầu về vệ sinh an toàn
5.1. Định tính
Tính tan Tan trong nước, rất ít tan trong etanol
Sắc ký bản mỏng Vết chính trong sắc ký đồ bản mỏng của dung dịch thử phải tương ứng về Rf, màu sắc và với vết chính trong sắc ký đồ bản mỏng của chất chuẩn.
5.2. Độ tinh khiết
Hàm lượng nước Không lớn hơn 7,0%
Tro sulfat Không lớn hơn 0,1%
Niken Không lớn hơn 2mg/kg
Chì Không lớn hơn 1mg/kg
Tổng kim loại nặng Không lớn hơn 10mg/kg
D-Manitol Không lớn hơn 3%
D-Sorbitol Không lớn hơn 6%
Đường thử Không lớn hơn 0,3%
5.3. Định lượng isomalt Không nhỏ hơn 98% hỗn hợp của mono- và disaccarid hydro hóa; Không nhỏ hơn 86% tổng C12H24O11 và C12H24O11.2H2O (tính theo chế phẩm khan).
6. Phương pháp thử
6.1. Thử định tính
Sắc ký bản mỏng
Bản mỏng:
Bản mỏng nền nhôm tráng sẵn kích cỡ khoảng 12cm, độ dày lớp hấp thụ khoảng 0,2mm Kieselgel 60 F254, art 5554, loại của Merk hoặc tương đương.
Dung dịch chuẩn:
Hòa tan 500mg mỗi loại đường sau đây trong 100ml nước cất:
- Sorbitol;
- Mannitol;
- Lactitol
- Maltitol
- 6-O-α-D-Glucopyranosyl-D-sorbitol (1,6-GPS)
- 1-O-α-D-Glucopyranosyl-D-manni-tol dihydrat (1,1-GPM)
Dung dịch mẫu thử:
Hoà tan 500mg mẫu thử isomalt trong 100ml nước cất
- Dung dịch A: Isopropanol: n-butanol: dung dịch acid boric trong nước cất (25mg/ml): acid acetic: acid propionic (50:30:20:2:16 - v:v:v:v)
- Dung dịch B: Etylacetat: pyridin: nước cất: acid acetic: acid propionic (50:50:10:5:5 - v:v:v:v)
- Dung dịch hiện màu:
A: Natri metaperiodat 0,1% trong nước cất (w/w)
B: etanol: acid sulfuric: anisandedyd: acid acetic (90:5:1:1 - v:v:v:v)
Tiến hành:
Chấm khoảng 0,3μl mỗi chất chuẩn và dung dịch mẫu thử vào vạch xuất phát của bản mỏng. Làm khô các điểm chấm. Khai triển sắc ký lên đến độ cao cách vạch xuất phát khoảng 10cm trong bình chứa dung môi A hoặc dung môi B. Đợi bản mỏng khô, nhúng bản mỏng vào dung dịch hiện màu A trong khoảng 3 giây.
Làm khô bản mỏng. Chú ý bản mỏng cần được làm khô hoàn toàn cả hai mặt. Nhúng bản mỏng vào dung dịch hiện màu B trong 3 giây và làm khô đến khi những vết màu có thể nhìn thấy được.
Giá trị Rf và những vết màu của các chất trên bản mỏng được liệt kê dưới đây:
Giá trị Rf
|
| Màu | Dung môi A | Dung môi B |
| Mannitol | đỏ sáng | 0,36 | 0,40 |
| Sorbitol | Nâu | 0,36 | 0,36 |
| GPM | Xanh - nâu | 0,28 | 0,16 |
| GPS | Xanh - nâu | 0,25 | 0,13 |
| Maltitol | Xanh lá cây | 0,26 | 0,22 |
| Lactitol | Xanh ô liu - xanh | 0,23 | 0,14 |
Giá trị Rf có thể biến đổi chút ít tuỳ thuộc vào nguồn cung cấp bản mỏng
Những vết chính trong sắc ký đồ thu được của dung dịch mẫu thử isomalt phải tương đương về giá trị Rf và màu sắc trong sắc ký của GPM và GPS.
6.2. Thử độ tinh khiết
Xác định hàm lượng nước
Phương pháp Karl-Fischer
Xem hướng dẫn chung các phương pháp thử (P.VC.03)
Xác định tro sulfat
Xem hướng dẫn chung các phương pháp thử (P.VC.11)
Chú ý: Lấy 5g mẫu thử
Xác định niken
Phương pháp quang phổ hấp thụ nguyên tử - kỹ thuật nguyên tử hóa ngọn lửa
Hóa chất:
Dung dịch amoni pyrolidindithio-cacbamat
Thiết bị:
Máy quang phổ hấp thụ nguyên tử: hoạt động theo các điều kiện sau:
- Vạch phổ đo: 232,0nm.
- Cường độ đèn: 70% Imax
- Khe đo: 0,5nm
- Tốc độ không khí nén: 4,5ml/phút
- Tốc độ axetylen: 1,2ml/phút
- Các điều kiện khác tuỳ theo yêu cầu riêng cho mỗi loại thiết bị
Dung dịch thử:
Hòa tan 20g mẫu thử vào hỗn hợp dung dịch acid acetic loãng TT.002 và nước cất (1:1) và định mức tới 100ml bằng hỗn hợp đó. Thêm 2ml dung dịch ammoni pyrolidin dithiocacbamat 1% và 10ml metyl isobutyl keton. Lắc, để yên cho tách lớp. Sử dụng lớp metyl isobutyl keton.
Dung dịch chuẩn:
Thêm 0,5ml; 1ml; 1,5ml dung dịch chuẩn nickel nồng độ 10mg/kg tương ứng vào 3 dung dịch chuẩn được chuẩn bị giống như dung dịch thử.
Tiến hành:
Sử dụng metyl isobutyl keton làm mẫu trắng, hiệu chỉnh máy về 0. Tiến hành đo mẫu tại điều kiện mô tả như trên.
Xác định chì
Xem hướng dẫn chung các phương pháp thử (P.VC.09)
Xác định tổng các kim loại nặng
Xem hướng dẫn chung các phương pháp thử (P.VC.10)
Chú ý: Lấy 2g mẫu thử
Xác định D-Mannitol
Xem Định lượng Isomalt
Xác định D-Sorbitol
Xem Định lượng Isomalt
Xác định đường thử
Xem hướng dẫn chung các phương pháp thử (P.HC.03)
Đánh giá kết quả:
Lượng Cu2O thu được không được vượt quá 50mg.
6.3. Định lượng isomalt
Thiết bị:
• Sắc ký khí: Detector ion hóa ngọn lửa; Cột sắc ký: Fused silica HT - 8(25m x 0,22mm x 0,25μm) hoặc tương đương; Nguồn cung cấp khí mang: heli
• Điều kiện sắc ký:
- Chương trình nhiệt độ: 800C (trong 3 phút); gia nhiệt 100C/phút đến 2100C; gia nhiệt tiếp 50C/phút đến 3500C, giữ trong 6 phút.
- Chương trình nhiệt độ buồng bơm mẫu: 300C; gia nhiệt 2700C/phút đến 3000C; giữ trong 49 phút.
- Tốc độ dòng ban đầu 1ml/phút tại 800C và 1 atm, tốc độ chia dòng 25ml/phút.
Dung dịch chuẩn nội:
Hòa tan một lượng cân thích hợp của phenyl-β-D-glucopyranosid và maltitol trong nước cất để thu được một dung dịch có nồng độ khoảng 1mg phenyl-β-D-glucopyranosid và 50mg maltitol trong 1g nước cất.
Dung dịch chuẩn:
Hoà tan một lượng chính xác 1-O-α-D-Glucopyranosyl-D-mannitol dihydrat (1,1-GPM) 6-O-α-D-Glucopyranosyl-D-sorbitol (1,6-GPS) trong nước cất để thu được 2 dung dịch riêng biệt có nồng độ 50mg/mỗi g dung dịch. Làm tương tự như vậy với một dung dịch chuẩn chứa khoảng 1mg mannitol/g và 1mg sorbitol/g.
Dung dịch mẫu thử:
Cân chính xác 1g mẫu thử Isomalt cần xác định hoà tan trong với nước cất để thu được dung dịch có nồng độ 10g/100g.
Tiến hành:
Lấy 100mg dung dịch chuẩn hoặc dung dịch mẫu thử cho vào ống thử thủy tinh có nút đậy và thêm 100mg dung dịch chuẩn nội. Làm mất nước bằng phương pháp làm khô lạnh và hòa tan cặn với 1ml pyridin. Thêm 4mg O-benzyl-hydroxylamin hydroclorid, đóng nắp ống thử, để yên trong 12 giờ ở nhiệt độ phòng. Thêm 1ml N-metyl-N-trimetylsilyl-trifluoroacetamid (MSTFA) và đun nóng đến 800C trong 12 giờ, thỉnh thoảng lắc và để nguội. Bơm chính xác 1μl các dung dịch này vào máy sắc ký khí đã được đặt ở điều kiện như trên với khí mang là He.
- Giá trị thời gian lưu:
Các monosaccarid hydro hóa:
Mannitol: 19,5 phút
Sorbitol: 19,6 phút
Dung dịch chuẩn nội: Phenyl-β-D-glucopyranosid: 26,8 phút
Matlitol: 33,5 phút
Các disaccarid hydro hóa: 32-36 phút
1,1 - GPS: 33,9 phút
1,1 - GPM: 34,5 phút
1,6 - GPS: 34,6 phút
Tính kết quả:
Tính phần trăm từng thành phần riêng rẽ, Wi trong mẫu thử cần xác định theo công thức sau:
Wi = x 100
Trong đó:
ai: diện tích pic của cấu tử I (μV.s)
as: diện tích pic của chất nội chuẩn (μV.s)
ms: khối lượng chất nội chuẩn sử dụng để dẫn xuất hóa (mg)
misomalt: khối lượng mẫu sử dụng để dẫn xuất hóa (mg)
Fi = fi / fs
fi: hệ số đáp ứng của cấu tử i; fi = (ai/mi) x (100/% tinh khiết)
fs: hệ số đáp ứng của chất nội chuẩn; fs = (as/ms) x (100/% tinh khiết)
Chú ý: Sử dụng maltitol làm chất nội chuẩn để tính lượng disaccarid hydro hóa (VD: 1,1-GPM; 1,6-GPS) và phenyl-α-D-glucosid là chất nội chuẩn để tính monosaccarid hydro hóa (sorbitol, mannitol). Đối với tổng lượng saccarid khác (đã hydro hóa hoặc chưa hydro hóa) lấy 100% trừ đi tỷ lệ của 1,1-GPM; 1,6-GPS; sorbitol và mannitol.
QUY ĐỊNH ĐÁNH GIÁ VỆ SINH AN TOÀN SUCRALOZA
1. Tên khác, ký hiệu 4,1’,6’-triclorogalactosucroza; Chỉ số INS: 955
2. Định danh
Tên hóa học 1,6-diclo-1,6-dideoxy-β-D-fructofuranosyl-4-clo-4-deoxy-α-D-galactopyranosid
Mã số C.A.S. 56038-13-2
Công thức hóa học C12H19Cl3O8
| Công thức phân tử |
|
Khối lượng phân tử 397,64
3. Mô tả ngoại quan Bột tinh thể trắng, không mùi, vị ngọt
4. Chức năng Chất tạo ngọt
5. Yêu cầu về vệ sinh an toàn
5.1. Định tính
Tính tan Tan tốt trong nước, metanol, etanol, ít tan trong etyl acetat
Phổ hấp thụ hồng ngoại Phổ hồng ngoại của chất trên nền chất mang KBr của mẫu thử phải có cực đại tại các số sóng tương ứng so với phổ hồng ngoại của chất chuẩn trong cùng điều kiện đo.
Kiểm tra sắc ký bản mỏng Vết chính trong sắc ký đồ bản mỏng của dung dịch thử phải có cùng giá trị Rf với vết chính trong sắc ký đồ bản mỏng của dung dịch chuẩn.
5.2. Độ tinh khiết
Góc quay cực riêng [α]D20: +84,00 tới +87,50 (tính theo chế phẩm khan)
Hàm lượng nước Không lớn hơn 2,0%
Tro sulfat Không lớn hơn 0,7%
Asen Không lớn hơn 3mg/kg
Tổng các kim loại nặng Không lớn hơn 10mg/kg
Các di-saccarid clo hoá khác Đạt (mô tả trong phần phương pháp thử)
Các mono-saccarid clo hóa Đạt (mô tả trong phần phương pháp thử)
Oxid triphenylphosphin Không lớn hơn 150mg/kg
Metanol Không lớn hơn 0,1%
5.3. Định lượng C12H19Cl3O8 Từ 98% đến 102% C12H19Cl3O8 (tính theo chế phẩm khan)
6. Phương pháp thử
6.1. Thử định tính
Phổ hấp thụ hồng ngoại
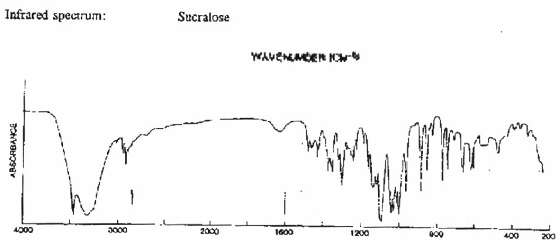
Sắc ký bản mỏng
Bản mỏng:
Sử dụng bản mỏng sắc ký pha ngược (VD: Whatman LCK18), chiều dày lớp silicagel hấp thụ là 0,2mm
Pha động:
Dung dịch NaCl 5% (m/v): CH3CN (7:3 - v:v)
Thuốc thử:
Dung dịch H2SO4 đặc trong metanol 15% (v:v)
Dung dịch chuẩn:
- Hòa tan 1g chuẩn trong 10ml metanol (dung dịch A)
- Pha loãng 0,5ml dung dịch A tới 100ml bằng metanol (dung dịch B)
Tiến hành:
- Hoà tan 1g mẫu thử trong 10ml metanol. Chấm 5μl dung dịch A, dung dịch B, dịch thử lên bản mỏng tại vạch xuất phát. Cho bản mỏng vào bình sắc ký đã để bão hòa dung môi. Để dung môi khai triển cho tới khi tiền tuyến dung môi cách vạch xuất phát 17cm. Lấy bản sắc ký ra khỏi bình, để khô và phun sương thuốc thử lên bản sắc ký. Sấy bản sắc ký tại 1250C trong 10 phút.
- Vết chính trong sắc ký đồ bản mỏng của dung dịch thử phải tương ứng về Rf, màu sắc và kích cỡ với vết chính trong sắc ký đồ bản mỏng của dung dịch A.
6.2. Thử độ tinh khiết
Xác định góc quay cực riêng
Xem hướng dẫn chung các phương pháp thử (P.VL.03)
Chú ý: Đo dung dịch mẫu thử nồng độ 10% (m/v) trong nước cất.
Xác định hàm lượng nước
Phương pháp chuẩn độ Karl-fischer
Xem hướng dẫn chung các phương pháp thử (P.VC.03)
Xác định tro sulfat
Xem hướng dẫn chung các phương pháp thử (P.VC.11)
Xác định asen
Xem hướng dẫn chung các phương pháp thử (P.VC.08)
Xác định tổng các kim loại nặng
Xem hướng dẫn chung các phương pháp thử (P.VC.10-Phương pháp 2)
Chú ý: Lấy 2g mẫu thử
Xác định các Disacarid clo hóa khác
Tiến hành như trong phần “kiểm tra sắc ký bản mỏng”
Các vết phụ trong sắc ký đồ dịch thử không được đậm hơn vết chính trong sắc ký đồ dung dịch B (vết chính có trị số Rf tương đương với vết chính trong sắc ký đồ của dung dịch A).
Xác định các Monosacarit clo hóa
Bản mỏng:
Dùng bản sắc ký lớp mỏng pha tĩnh Merck silicagel 60 hoặc tương đương có độ dày lớp pha tĩnh 0,25mm
Thuốc thử phun sương:
Hòa tan 1,23g p-anisidin và 1,66g phtalic acid trong 100ml metanol. Giữ dung dịch trong chỗ tối và trong tủ lạnh để ngăn dung dịch mất màu. Loại bỏ dung dịch nếu dung dịch mất màu.
(Chú ý: p-anisidin là chất độc hấp thụ qua da và qua đường hô hấp nên sử dụng phải cẩn thận).
Dung dịch chuẩn A:
Cân (chính xác tới 0,001g) 10,0g mannitol: Hòa tan vào trong bình định mức 100ml và định mức tới vạch bằng nước cất.
Dung dịch chuẩn B:
Hoà tan 10g mannitol và 40mg fructoza (Loại dùng phân tích) trong bình định mức 100ml, định mức tới vạch bằng nước cất.
Dịch thử:
Hoà tan 5g mẫu trong 5ml metanol cho vào bình định mức 10ml và định mức tới vạch bằng metanol
Tiến hành:
Chấm 5μl dung dịch A và B vào trên bản sắc ký. Dung dịch được chấm từ từ từng μl, làm khô rồi lại chấm tiếp. Chấm 5μl dung dịch mẫu trên bản sắc ký theo cách tương tự như trên, ba điểm chấm có kích thước tương tự như nhau. Phun sương bản mỏng với dung dịch thuốc thử phun sương và sấy ở 1000C ± 20C trong 15 phút. Ngay sau khi sấy, quan sát bản mỏng trên nền tối. Vết dung dịch mẫu không được đậm màu hơn vết dung dịch B (tương ứng với giới hạn của tổng monosaccarid clo hóa tối đa 0,1%) (Nếu vết mannitol dung dịch chuẩn A có màu sẫm đen cho thấy thử nghiệm không đạt do thời gian sấy bản mỏng sắc ký quá lâu, cần phải tiến hành lại thử nghiệm).
Xác định Oxid Triphenyl phosphin
Phương pháp sắc ký lỏng phân giải cao
Hệ thống sắc ký:
Loại sắc ký lỏng phân giải cao, điều khiển ở nhiệt độ phòng. Pha tĩnh là cột pha ngược C18 (10cm x 8mm) Pha động được duy trì ở tốc độ dòng là 1,5ml/phút, detector UV đo tại bước sóng 220nm.
Pha động:
CH3CN: H2O (67:33 - v:v) đã lọc trên màng lọc kích thước lỗ lọc 0,45μm, đã đuổi khí.
Dung dịch chuẩn:
Cân chính xác 100mg oxid triphenyl-phosphin vào trong bình thể tích 10ml. Hoà tan và định mức tới thể tích bằng dung môi pha động. Lấy 1,0ml dung dịch này cho vào bình 100ml và định mức tới 100ml bằng pha động. Từ dung dịch chuẩn bị được pha loãng với pha động 100 lần được dung dịch chuẩn làm việc. Dung dịch chuẩn làm việc được lọc trân màng thước lỗ 0,45μm
Dịch thử:
Cân chính xác khoảng 100mg mẫu cho vào trong bình thể tích 10ml. Hoà tan và định mức tới vạch mức bằng pha động. Dịch thử được lọc trân màng thước lỗ 0,45μm.
Tiến hành:
Bơm 25μl dung dịch chuẩn làm việc và dung dịch thử nghiệm vào trong máy sắc ký (mỗi dịch bơm hai lần). Thời gian lưu với triphenylphosphin oxid là 6 phút. Ghi lại diện tích trung bình của peak với dung dịch chuẩn và dung dịch thử nghiệm là As và At tương ứng. Nồng độ của oxid triphenylphosphin (OTPP) trong mẫu được tính theo công thức sau:
OTPP (mg/kg) =
Xác định metanol
Thiết bị:
Máy sắc ký khí với detector ion hóa ngọn lửa (FID), cột thủy tinh kích thước 2,1m x 4,0mm (đường kính trong), được nhồi pha tĩnh Porapak PS cỡ hạt 80-100 mesh hoặc vật liệu tương đương.
Điều kiện làm việc: Điều kiện hoạt động có thể biến đổi tuỳ thuộc vào dụng cụ sử dụng riêng nhưng với hệ sắc ký này có thể đặt điều kiện:
Nhiệt độ cột: 1500C (đẳng nhiệt)
Nhiệt độ buồng bơm: 2000C
Nhiệt độ detector: 2500C
Khí mang nitơ: Tốc độ dòng 20ml/phút
Dung dịch chuẩn:
Dùng pipet có bầu, lấy 2,0ml metanol vào trong bình thủy tinh 100ml, pha loãng tới mức với pyridin và trộn đều hỗn hợp. Chuyển 11,0ml dung dịch này vào bình thể tích 100ml và pha loãng tới mức với pyridin và trộn đều hỗn hợp.
Dung dịch mẫu:
Cân chính xác khoảng 2g mẫu vào trong bình định mức thể tích 10ml pha loãng tới mức với pyridin và trộn đều hỗn hợp.
Tiến hành:
Bơm 1μl dung dịch chuẩn vào trong cột sắc ký khí, bơm 3 lần, độ lệch chuẩn về diện tích pic giữa các lần bơm lặp lại không lớn hơn 2,0%. Tính diện tích peak trung bình của dung dịch chuẩn. Tương tự tiêm 1μl, bơm 3 lần.
Tính kết quả:
Tính diện tích trung bình của peak và xác định nồng độ metanol theo công thức sau đây:
MeOH (%) =
Trong đó:
- Sa: diện tích peak của mẫu
- Cs: nồng độ của metanol trong dung dịch chuẩn (%) (thể tích của metanol x hệ số pha loãng x tỷ trọng của dung môi bằng 2 x 104 x 0,79 x 100).
- Vs: thể tích dung dịch mẫu
- As: diện tích peak của dung dịch mẫu
- Ws: khối lượng của mẫu
6.3. Định lượng sucraloza
Phương pháp sắc ký lỏng phân giải cao
Máy sắc ký:
Sắc ký lỏng phân giải cao hoạt động ở nhiệt độ phòng, cột pha ngược C18 kích thước 10cm, 5μm. Pha động được duy trì tốc độ dòng 1,5ml/ph. Detector UV đo ở bước sóng 190nm hoặc sử dụng detector đo chiết suất.
Pha động:
CH3CN:H2O (15:85 - v:v), lọc qua màng có kích thước lỗ lọc 0,45μm, đã đuổi khí.
Dung dịch chuẩn:
Cân khoảng 250mg sucraloza, cho vào bình định mức 25ml. Hòa tan và định mức tới 25ml bằng pha động. Lọc dung dịch qua màng lọc kích thước lỗ 0,45μm. Ghi lại lượng cân chính xác (Ws).
Dung dịch thử:
Cân khoảng 250mg mẫu cho vào trong bình định mức 25ml. Hoà tan và định mức tới vạch bằng pha động. Lọc qua màng lọc kích thước lỗ 0,45μm. Ghi lại khối lượng mẫu chính xác (Wt).
Bơm chuẩn và thử hệ thống:
Bơm 20μl dung dịch chuẩn (3 lần). Thời gian lưu của sucraloza sẽ là khoảng 9 phút (nếu sử dụng cột Rad-Pak C18 thời gian lưu là ~10 phút, nếu sử dụng cột khác học cột có kích thước khác, cần nghiên cứu điều chỉnh tỷ lệ CH3CN để thu được pic có thời gian lưu đã định). Tổng các hệ số sai lệch (100 x độ lệch chuẩn chia cho diện tích trung bình của peak), của các giá trị diện tích pic không vượt quá 2%.
Tiến hành
Bơm 20μl dịch thử (bơm 3 lần). Ghi lại giá trị diện tích pic trung bình của 3 lần bơm.
Tính kết quả:
Tính % hàm lượng theo công thức dưới đây:
Hàm lượng (%) =
Trong đó:
At: diện tích pic của mẫu thử
As: diện tích pic của sucraloza chuẩn
Wt: khối lượng mẫu thử
Ws: khối lượng sucraloza chuẩn
Tính % độ tinh khiết đối với chế phẩm khan và không có metanol sử dụng giá trị thu được từ phép thử hàm lượng nước và metanol.
QUY ĐỊNH ĐÁNH GIÁ VỆ SINH AN TOÀN RED 2G
1. Tên khác, ký hiệu CI Food Red 10, azogeramin; Chỉ số INS: 128; Chỉ số EEC: E218
2. Định danh Red 2G chủ yếu gồm dinatri8-acetamido-1-hydroxy-2-phenylazo-3,6-naphtalensulfonat và các chất màu phụ cùng với NaCl hoặc NaSO4 là những thành phần không màu chính.
Nhóm chất màu Bisazo
Tên hoá học Dinatri 8-acetamit-1-hydroxy-2-phenylazo-3,6-naphtalensulfonat
Mã số C.A.S. CI(1975) số 18050; Số CAS 3734-67-6
Công thức hóa học C18H13N3Na2O8S2
| Công thức phân tử |
|
Khối lượng phân tử 509,43
3. Mô tả ngoại quan Dạng bột hoặc hạt nhỏ màu đỏ
4. Chức năng Phẩm màu thực phẩm
5. Yêu cầu về vệ sinh an toàn
5.1. Định tính
Tính tan Tan trong nước, không tan trong etanol
Định tính các chất màu Đạt (mô tả trong phần phương pháp thử)
5.2. Độ tinh khiết
| Giảm khối lượng khi làm khô Cl- và SO42- tính theo muối natri |
| Không lớn hơn 20% |
Các chất không tan trong nước Không lớn hơn 0,2%
Asen Không lớn hơn 3mg/kg
Chì Không lớn hơn 10mg/kg
Tổng các kim loại nặng Không lớn hơn 40mg/kg
Các chất màu phụ Không lớn hơn 2%
Các chất hữu cơ ngoài chất màu
| - Acid 5-acetamido-4-hydroxy-2,7-naphtalen-2,7-disulfonic - Acid 5-amino-4-hydroxy-2,7-naphtalen-2,7-disulfonic |
| Tổng hàm lượng: Không lớn hơn 0,3% | |
| Các amin thơm bậc nhất không sulfonat hóa | Không lớn hơn 0,01% (tính theo anilin) | ||
| Hàm lượng các chất có thể chiết bằng ete | Không lớn hơn 0,2% | ||
5.3. Định lượng chất màu chính Không nhỏ hơn 80% tổng chất màu
6. Phương pháp thử
6.1. Thử định tính
Thử định tính các chất màu
Xem hướng dẫn chung các phương pháp thử (P.PM.01)
Chú ý: Các vết trên sắc ký đồ bản mỏng của mẫu thử phải tương ứng về giá trị Rf và màu sắc với sắc ký đồ bản mỏng của chất chuẩn khi tiến hành trong cùng một điều kiện sắc ký.
6.2. Thử độ tinh khiết
Xác định giảm khối lượng khi làm khô
Xem Hướng dẫn chung các phương pháp thử (P.PM.02)
Chú ý: Mẫu được sấy tại 1350C đến khối lượng không đổi.
Xác định Cٱ và SO42ٱ tính theo muối natri
Xem hướng dẫn chung các phương pháp thử (P.PM.03 và P.PM.04)
Xác định các chất không tan trong nước
Xem hướng dẫn chung các phương pháp thử (P.PM.05)
Xác định asen
Xem hướng dẫn chung các phương pháp thử (P.VC.09)
Xác định chì
Xem hướng dẫn chung các phương pháp thử (P.PM.09)
Xác định tổng các kim loại nặng
Xem hướng dẫn chung các phương pháp thử (P.PM.10-Phương pháp 2)
Xác định các chất màu phụ
Xem hướng dẫn chung các phương pháp thử (P.PM.06)
Chú ý: Điều kiện tiến hành sắc ký:
- Hệ dung môi khai triển: Hệ số 4.
Khoảng cách giữa tiền tuyến dung môi và vạch xuất phát ~ 17cm
Xác định các chất hữu cơ ngoài chất màu
Xem hướng dẫn chung các phương pháp thử (P.PM.07)
Chú ý: Điều kiện cho HPLC rửa giải Gradient: thành phần dung môi 1%-100%, tốc độ thay đổi 2,5%/phút (tuyến tính)
Xác định các amin thơm bậc nhất không sulfonat hóa
Xem hướng dẫn chung các phương pháp thử (P.PM.09)
Xác định các chất có thể chiết bằng ete
Xem hướng dẫn chung các phương pháp thử (P.PM.08)
6.3. Định lượng chất màu chính
Phương pháp chuẩn độ với Titan clorid
Xem hướng dẫn chung các phương pháp thử (P.PM.11)
Chú ý:
- Cân 1,4 - 1,5g mẫu
- Đệm: 15g Natri hydro tartrat
- 1ml TiCl3 0,1N tương đương với 28,83g các chất màu chính
QUY ĐỊNH ĐÁNH GIÁ VỆ SINH AN TOÀN GREEN S
1. Tên khác, ký hiệu CI food green 4, food green S; Chỉ số INS: 142; Chỉ số EEC: E142
2. Định danh Green S chủ yếu gồm natri N-[4-] [4-(dimetylamino) phenyl] (2-hydroxy-3,6 disulfo-1-naphtalenyl) metylen]-2,5-cyclohexadien-1-ylldenel-N-metylme-tanaminin và các chất màu phụ cùng với NaCl và (hoặc) Na2SO4 là các thành phần không màu chính.
Nhóm chất màu Triarylmetan
Tên hóa học Natri N-[4-] [4-(dimetylamino) phenyl] (2-hydroxy-3,6 disulfo-1-naphtalenyl) metylen]-2,5-cyclohexadien-1-ylldenel-N-metylmetanaminin; hoặc Natri 5-[4-dimetylamino-α-(4-dimetylimini-ocyclohexa-2,5-dienyliden) benzyl] 6-hydroxy-7-sulfonat-naphtalen-2-sulfonat
Mã số C.A.S Số CI(1975): 44090; Số CAS: 860-22-0
Công thức hóa học C27H25N2NaO7S2
| Công thức phân tử |
|
Khối lượng phân tử 576,63
3. Mô tả ngoại quan Dạng bột hoặc hạt màu xanh đậm
4. Chức năng Phẩm màu thực phẩm
5. Yêu cầu về vệ sinh an toàn
5.1. Định tính
Tính tan Tan trong nước, ít tan trong etanol
Định tính các chất màu Đạt (mô tả trong phần phương pháp thử)
5.2. Độ tinh khiết
Giảm khối lượng khi làm khô
Cl- và SO42- tính theo muối natri Không lớn hơn 20%
Các chất không tan trong nước Không lớn hơn 0,2%
Asen Không lớn hơn 3mg/kg
Chì Không lớn hơn 10mg/kg
Crôm Không lớn hơn 50mg/kg
Tổng các kim loại nặng Không lớn hơn 40mg/kg
Các chất màu phụ Không lớn hơn 1%.
Các chất hữu cơ ngoài chất màu
| - 4,4’-bis(dimetylamino)benzhydryl alcol | Không lớn hơn 0,1% |
| - 4,4’-bis(dimetylamino)benzo-phenon | Không lớn hơn 0,1% |
| - 3-Hydroxynaphtalen-2,7-disulfonic acid | Không lớn hơn 0,2% |
| - Các amin thơm bậc nhất không sulfonat hóa | Không lớn hơn 0,01% tính theo anilin |
Leuco base Không lớn hơn 5%
Các chất có thể chiết bằng ete Không lớn hơn 0,2%
5.3. Định lượng chất màu chính Không nhỏ hơn 80% tổng các chất màu
6. Phương pháp thử
6.1. Thử định tính
Định tính các chất màu
Xem hướng dẫn chung các phương pháp thử (P.PM.01)
Chú ý: Các vết trên sắc ký đồ bản mỏng của mẫu thử phải tương ứng về giá trị Rf và màu sắc với sắc ký đồ bản mỏng của chất chuẩn khi tiến hành trong cùng một điều kiện sắc ký.
6.2. Thử độ tinh khiết
Xác định giảm khối lượng khi làm khô
Xem hướng dẫn chung các phương pháp thử (P.PM.02)
Chú ý: Mẫu được sấy tại 1350C đến khối lượng không đổi
Xác định Cl- và SO42- tính theo muối natri
Xem hướng dẫn chung các phương pháp thử (P.PM.03 và P.PM.04)
Xác định các chất không tan trong nước
Xem hướng dẫn chung các phương pháp thử (P.PM.05)
Xác định asen
Xem hướng dẫn chung các phương pháp thử (P.VC.09)
Xác định chì
Xem hướng dẫn chung các phương pháp thử (P.VC.09)
Xác định Crôm
Xem hướng dẫn chung các phương pháp thử (P.VC.09)
Xác định tổng các kim loại nặng
Xem hướng dẫn chung các phương pháp thử (P.VC.09-Phương pháp 2)
Xác định các chất màu phụ
Xem hướng dẫn chung các phương pháp thử (P.PM.06)
Chú ý: Điều kiện tiến hành sắc ký:
- Hệ dung môi khai triển: Hệ số 2
- Khoảng cách giữa tiền tuyến dung môi và vạch xuất phát ~ 17cm
Xác định các hợp chất hữu cơ ngoài chất màu
Xem hướng dẫn chung các phương pháp thử (P.PM.07)
Chú ý: Điều kiện cho HPLC rửa giải Gradient: thành phần dung môi 2% - 100%, tốc độ thay đổi 2,0%/phút (tuyến tính).
Xác định leuco base
Xem hướng dẫn chung các phương pháp thử (P.PM.10)
Chú ý:
- Cân 110 ± 5mg mẫu thử.
- Độ hấp thụ riêng:
(a) = 0,1725/mg/L/cm tại bước sóng 634nm;
Tỷ lệ = 0,9600
Xác định các chất có thể chiết bằng ete
Xem hướng dẫn chung các phương pháp thử (P.PM.08).
6.3. Định lượng chất màu chính
Phương pháp chuẩn độ bằng Titan clorid
Xem hướng dẫn chung các phương pháp thử (P.PM.11)
Chú ý:
- Cân 1,4 - 1,5g mẫu
- Đệm: 15g Natri hydro tartrat
1ml TiCl3 0,1N tương đương với 28,83mg các chất màu chính
QUY ĐỊNH ĐÁNH GIÁ VỆ SINH AN TOÀN BRILLIANT BLACK BN
1. Tên khác, ký hiệu CI Food Black 1, Black BN; Chỉ số INS: 151; Chỉ số EEC: E151
2. Định danh Brilliant black BN chủ yếu gồm tetranatri 4-acetamido-5-hydroxy-6-[7-sulfonat-4-(4-sulfonat-phenylazo)-1-naphtylazo]-1,7-naphtalen-disulfonat và một số chất Màu phụ cùng với NaCl và (hoặc) Na2SO4 là những thành phần không màu chính.
Nhóm chất màu Bisazo
Tên hóa học Tetranatri 4-acetamido-5-hydroxy-6-[7-sulfonat-4-(4-sulfonat-phenylazo)-1-naphtylazo]-1,7-naphtalen-disulfonat
Mã số CI (975) số 28840; CAS 2519-30-4
Công thức hóa học C28H17N5Na4O14S4
Công thức phân tử
Khối lượng phân tử 867,69
3. Mô tả ngoại quan Dạng bột hoặc dạng hạt màu đen
4. Chức năng Phẩm màu thực phẩm
5. Yêu cầu về vệ sinh an toàn
5.1. Định tính
Tính tan Tan trong nước, ít tan trong etanol
Định tính các chất màu Đạt (mô tả trong phần phương pháp thử).
5.2. Độ tinh khiết
| Giảm khối lượng khi làm khô Cl- và SO42- tính theo muối natri | Không lớn hơn 20% |
Các chất không tan trong nước Không lớn hơn 0,2%
Asen Không lớn hơn 3mg/kg
Chì Không lớn hơn 10mg/kg
Tổng các kim loại nặng Không lớn hơn 40mg/kg
Các chất màu phụ Không lớn hơn 4%
Các chất hữu cơ ngoài chất màu
| - acid 4-acetamid-5-hydroxy-1,7-naphtalendisulfonic; - acid 4-amin-5-hydroxy-1,7-naphtalendisulfonic; - acid 8-amin-2-naphtalensulfonic; acid sulfanilic; - 4,4’-Diazoamin-di (acid bezensulfuric) | Tổng hàm lượng không lớn hơn 0,8% | |
| - Các amin thơm bậc nhất không sulfonat hóa | Không lớn hơn 0,01% (tính theo anilin) | |
Các chất có thể chiết bằng ete Không lớn hơn 0,2%
5.3. Định lượng chất màu chính Không nhỏ hơn 80% tổng chất màu
6. Phương pháp thử
6.1. Thử định tính
Định tính các chất màu
Xem hướng dẫn chung các phương pháp thử (P.PM.01)
Chú ý: các vết trên sắc ký đồ bản mỏng của mẫu thử phải tương ứng về giá trị Rf và màu sắc với sắc ký đồ bản mỏng của chất chuẩn khi tiến hành trong cùng một điều kiện sắc ký.
6.2. Thử độ tinh khiết
Xác định giảm khối lượng khi làm khô
Xem hướng dẫn chung các phương pháp thử (P.PM.02)
Ghi chú: Mẫu thử được sấy tại 1350C tới trọng lượng không đổi.
Xác định Cl- và SO42- tính theo muối natri
Xem hướng dẫn chung các phương pháp thử (P.PM.03 và P.PM.04)
Xác định các chất không tan trong nước
Xem hướng dẫn chung các phương pháp thử (P.PM.05)
Xác định asen
Xem hướng dẫn chung các phương pháp thử (P.VC.09)
Xác định chì
Xem hướng dẫn chung các phương pháp thử (P.VC.09)
Xác định tổng các kim loại nặng
Xem hướng dẫn chung các phương pháp thử (P.VC.10-Phương pháp 2)
Xác định các chất màu phụ
Xem hướng dẫn chung các phương pháp thử (P.PM.06)
Chú ý:
- Dung môi khai triển sắc ký: Sắc ký (i): Hệ dung môi số 1
Sắc ký (ii): Hệ dung môi số 4.
- Khoảng cách từ vạch xuất phát đến tiền tuyến dung môi ~ 17cm.
Xác định các chất hữu cơ ngoài chất màu
Xem hướng dẫn chung các phương pháp thử (P.PM.07)
Chú ý:
Điều kiện HPLC: rửa giải Gradient: 2 đến 100% tốc độ 2%/phút (tuyến tính).
Xác định các amin thơm bậc nhất không sulfonat hóa.
Xem hướng dẫn chung các phương pháp thử (P.PM.09)
Xác định các chất có thể chiết bằng ete
Xem hướng dẫn chung các phương pháp thử (P.PM.08)
6.3. Định lượng chất màu chính
Phương pháp chuẩn độ với Titan clorid
Xem hướng dẫn chung các phương pháp thử (P.PM.11)
Chú ý:
- Cân 0,6 - 0,7g mẫu
- Đệm: 15g Natri hydro tartrat
- 1ml TiCl3 0,1N tương đương với 10,86mg các chất màu chính
QUY ĐỊNH ĐÁNH GIÁ VỆ SINH AN TOÀN BROWN HT
1. Tên khác, ký hiệu CI Food Brown 3, Chocolat brown HT; Chỉ số INS: 155; Chỉ số EEC: E155
2. Định danh Brown HT chủ yếu gồm dinatri 4,4-(2,4-dihydroxy-5-hydroxymetyl-1,3-phenylen-bisazo)di-1-naphtalensulfonat và những chất màu phụ cùng NaCl và (hoặc) Na2SO4 là những thành phần không màu chính.
Loại chất màu Bisazo
Tên hóa học Dinatri 4,4-(2,4-dihydroxy-5-hydroxymetyl-1,3-phenylen-bisazo di-1-naphtalensulfonat
Mã số C.A.S. CI(1975) số 20285; Số CAS 4553-89-3
Công thức hóa học C17H18N4Na2O9S2
Công thức phân tử
Khối lượng phân tử 652,57
3. Mô tả ngoại quan Dạng bột màu nâu hoặc hạt nhỏ
4. Chức năng Phẩm màu thực phẩm
5. Yêu cầu về vệ sinh an toàn
5.1. Định tính
Tính tan Tan trong nước, không tan trong etanol
Định tính các chất màu Đạt (mô tả trong phần phương pháp thử).
5.2. Độ tinh khiết
| Giảm khối lượng khi làm khô Cl- và SO42- tính theo muối natri | Không lớn hơn 30% |
Các chất không tan trong nước Không lớn hơn 0,2%
Asen Không lớn hơn 3mg/kg
Chì Không lớn hơn 10mg/kg
Tổng các kim loại nặng Không lớn hơn 40mg/kg
Các chất màu phụ Không lớn hơn 10%
Các chất hữu cơ ngoài chất màu
| - acid 4-amino naphtalen-1-sulfonic | Không lớn hơn 0,7% |
| - Các amin thơm bậc nhất không sulfonat hóa | Không lớn hơn 0,01%, tính theo anilin |
Các chất có thể chiết bằng ete Không lớn hơn 0,2%
5.3. Định lượng chất tạo chính Không nhỏ hơn 70% tổng chất màu
6. Phương pháp thử
6.1. Thử định tính
Thử định tính các chất màu
Xem hướng dẫn chung các phương pháp thử (P.PM.01)
Chú ý: các vết trên sắc ký đồ bản mỏng của mẫu thử phải tương ứng về giá trị Rf và màu sắc với sắc ký đồ bản mỏng của chất chuẩn khi tiến hành trong cùng một điều kiện sắc ký.
6.2. Thử độ tinh khiết
Giảm khối lượng khi làm khô
Xem hướng dẫn chung các phương pháp thử (P.PM.02)
Ghi chú: Sấy mẫu thử ở nhiệt độ 1350C đến khối lượng không đổi.
Xác định Cl- và SO42- (tính theo muối natri)
Xem hướng dẫn chung các phương pháp thử (P.PM.03 và P.PM.04)
Xác định các chất không tan trong nước
Xem hướng dẫn chung các phương pháp thử (P.PM.05)
Xác định asen
Xem hướng dẫn chung các phương pháp thử (P.VC.09)
Xác định chì
Xem hướng dẫn chung các phương pháp thử (P.VC.09)
Xác định tổng các kim loại nặng
Xem hướng dẫn chung các phương pháp thử (P.VC.10-Phương pháp 2)
Xác định các chất màu phụ
Xem hướng dẫn chung các phương pháp thử (P.PM.06)
Chú ý:
- Chuẩn bị dung dịch chuẩn: Hòa tan 1ml dung dịch chuẩn với 100ml nước cất, lắc đều. Lấy 0,1ml dung dịch này vào ống nghiệm, thêm 5ml dung dịch Aceton: nước (1:1-v:v), Thêm 14,9ml NaHCO3 0,05N, lắc đều. Đo độ hấp thụ quang của dung dịch chuẩn này.
- Dung môi khai triển sắc ký: Hệ dung môi số 6.
Thời gian khai triển sắc ký: ~ 14 giờ
Xác định các chất hữu cơ ngoài chất màu
Xem hướng dẫn chung các phương pháp thử (P.PM.07)
Chú ý:
- Điều kiện HPLC: rửa giải Gradient: 1 đến 100% (tốc độ 2%/phút).
Các amin thơm bậc nhất không sulfonat hóa (P.PM.09)
Xem hướng dẫn chung các phương pháp thử
Các chất có thể chiết bằng ete (P.PM.08)
Xem hướng dẫn chung các phương pháp thử
6.3. Định lượng chất màu chính
Phương pháp Quang phổ
Xem hướng dẫn chung các phương pháp thử (P.PM.11)
Chú ý:
Điều kiện:
Dung môi: Dung dịch đệm phosphat pH 7.
Pha loãng dung dịch A: từ 10ml đến 250ml
Độ hấp thụ riêng (a): 40,3
Bước sóng hấp thụ cực đại: 460nm.
HƯỚNG DẪN CHUNG CÁC PHƯƠNG PHÁP THỬ
HƯỚNG DẪN PHƯƠNG PHÁP THỬ CÁC TÍNH CHẤT VẬT LÝ: P.VL.
XÁC ĐỊNH KHOẢNG NHIỆT ĐỘ NÓNG CHẢY: P.VL.01
Khoảng nhiệt độ nóng chảy (gọi tắt là khoảng chảy) của một chất là khoảng nhiệt độ đã hiệu chỉnh, kể từ khi chất rắn bắt đầu nóng chảy và xuất hiện những giọt chất lỏng đầu tiên, đến khi chất rắn chuyển hoàn toàn sang trạng thái lỏng. Nhiệt độ nóng chảy (gọi tắt là điểm chảy) của một chất là nhiệt độ đã hiệu chỉnh, tại đó hạt chất rắn cuối cùng của chất thử nghiệm chuyển thành trạng thái lỏng, bắt đầu biến màu, hóa than hoặc sủi bọt.
Khi phải xác định khoảng chảy, nếu nhiệt độ bắt đầu hoặc nhiệt độ kết thúc nóng chảy không xác định rõ ràng, ta có thể chỉ xác định nhiệt độ kết thúc, hoặc nhiệt độ bắt đầu nóng chảy. Nhiệt độ này phải nằm trong giới hạn quy định trong chuyên luận riêng.
Để việc định tính hoặc thử tinh khiết của mẫu thử được tốt hơn, cần tiến hành xác định khoảng chảy của một hỗn hợp gồm các phần bằng nhau của mẫu thử và của mẫu đối chiếu (mẫu chuẩn). Nếu mẫu thử và mẫu đối chiếu khác nhau, hoặc nếu mẫu thử có lẫn tạp chất thì hỗn hợp sẽ bắt đầu nóng chảy ở nhiệt độ thấp hơn, và khoảng chảy sẽ rộng hơn quy định.
Điểm eutectic là nhiệt độ đã hiệu chỉnh, tại đó một hỗn hợp hai thành phần bắt đầu nóng chảy. Điểm eutectic không phụ thuộc vào tỷ lệ thành phần của hỗn hợp thử và đối với nhiều chất, cũng hằng định như điểm chảy.
Muốn xác định điểm eutectic, ta nghiền bằng cách thích hợp, mỗi hỗn hợp thành phần bằng nhau của chất thử và chất chỉ dẫn trong chuyên luận, sau đó tiến hành xác định nhiệt độ bắt đầu nóng chảy của hỗn hợp đó.
Để xác định khoảng chảy và điểm chảy, tùy theo tính chất lý học của từng chất, áp dụng một trong các phương pháp 1, hay phương pháp 2.
Phương pháp 1 (áp dụng cho các chất rắn dễ nghiền nhỏ)
Dụng cụ
Một bình thủy tinh chịu nhiệt, trong bình chứa một chất lỏng thích hợp, thường dùng là dầu parafin, hoặc để xác định ở nhiệt độ cao dùng dầu silicon, lượng chất lỏng đủ để nhúng chìm được nhiệt kế và mẫu thử sao cho bầu thủy ngân cách đáy bình 2cm.
Một dụng cụ khuấy có khả năng duy trì sự đồng nhất về nhiệt độ trong chất lỏng.
Một nhiệt kế đã được hiệu chuẩn và chia độ đến 0,50C, có khoảng nhiệt độ đo từ thấp nhất đến cao nhất không được quá 1000C. Ống mao quản, hàn kín một đầu, dài 6 - 8cm, đường kính trong 1,0 ± 0,1mm, thành ống dày khoảng 0,10 - 0,15mm.
Nguồn nhiệt, có thể điều chỉnh được.
Dụng cụ có thể được hiệu chuẩn với các chất chuẩn có điểm chảy được chứng nhận của Tổ chức y tế thế giới hay những chất thích hợp khác.
Cách xác định
Nghiền thành bột mịn chất thử đã làm khô 24 giờ trong bình với chất hút ẩm thích hợp, hoặc sấy khô 2 giờ ở 1000C - 1050C, trừ trường hợp có chỉ dẫn riêng trong chuyên luận.
Cho bột vào ống mao quản, lèn bột bằng cách gõ nhẹ ống mao quản xuống mặt phẳng cứng để có một lớp mẫu thử cao 4mm - 6mm. Giữ ống mao quản trong bình hút ẩm trước khi tiến hành xác định.
- Đun nóng bình đựng chất lỏng đến khi nhiệt độ thấp hơn điểm chảy dự kiến của chất thử khoảng 1000C, điều chỉnh nhiệt độ sao cho nhiệt độ tăng 10C trong 1 phút (trừ trường hợp có chỉ dẫn trong chuyên luận riêng), hoặc cho nhiệt độ tăng 30C trong 1 phút khi thử các chất không bền vì nhiệt.
Khi nhiệt độ đạt thấp hơn điểm chảy dự kiến khoảng 50C, lấy nhiệt kế ra, nhanh chóng buộc ống mao quản có mẫu thử vào nhiệt kế, sao cho lớp mẫu thử ngang với phần giữa bầu thủy ngân ở nhiệt kế. Đặt lại nhiệt kế vào bình.
Nhiệt độ mà tại đó nhìn thấy cột chất thử xẹp xuống, so sánh với một điểm nào đó trên thành ống, được xác định là điểm bắt đầu nóng chảy và nhiệt độ mà tại đó chất thử trở thành chất lỏng hoàn toàn, được xác định là điểm cuối của sự chảy hay điểm chảy. Tiến hành xác định ít nhất 3 lần. Các kết quả đo được phải không chênh lệch quá 10C thì tiến hành đo thêm 2 lần nữa và lấy trung bình của 5 lần thử nghiệm.
Hiệu chỉnh nhiệt độ quan sát được nếu nhiệt kế đo có sai số với nhiệt kế chuẩn và sự khác biệt nếu có giữa nhiệt độ của đoạn cột thuỷ ngân ngoài chất lỏng trong điều kiện hiệu chuẩn. Nhiệt độ của đoạn cột thủy ngân ở ngoài chất lỏng được xác định bằng cách đặt bầu thủy ngân của nhiệt kế phụ ở điểm giữa của phần cột thuỷ ngân lộ ra ngoài chất lỏng của nhiệt kế chính.
Tính nhiệt độ đã hiệu chỉnh theo công thức sau:
Thiệu chỉnh = T + 0,00015 N (Ts - t)
Trong đó:
T: Nhiệt độ đọc trên nhiệt kế chính
Ts: Nhiệt độ trung bình của đoạn cột thuỷ ngân ở ngoài chất lỏng của nhiệt kế chính trong điều kiện chuẩn hóa
t: Nhiệt độ của đoạn cột thuỷ ngân ở ngoài chất lỏng đọc trên nhiệt kế phụ tại điểm chảy.
N: Số khoảng chia độ (0C) của đoạn cột thuỷ ngân của nhiệt kế chính ở ngoài chất lỏng.
Phương pháp 2 (áp dụng cho các chất không nghiền thành bột được như mỡ, sáp, acid béo, parafin rắn, lanolin và chất tương tự, không tan trong nước).
Dụng cụ
Dùng dụng cụ như phương pháp 1, riêng ống mao quản hở hai đầu, dài 80 - 100mm, đường kính trong 0,8 - 1,2mm, thành dày 0,1 - 0,3mm.
Cách xác định
Cẩn thận làm chảy chất thử ở nhiệt độ thấp tối thiểu. Cắm ống mao quản vào chất thử sao cho lấy được một cột chất thử cao 10mm không có bọt. Dùng khăn sạch lau sạch phía ngoài ống mao quản, để nguội ở 100C hay thấp hơn trong 24 giờ, hoặc để tiếp xúc với nước đá ít nhất 2 giờ.
Đổ nước cất vào cốc thủy tinh đến chiều cao 6cm. Treo nhiệt kế vào giá và nhúng vào chính giữa cốc nước. Đun nóng cốc nước đến nhiệt độ thấp hơn điểm chảy dự kiến 100C, lấy nhiệt kế ra nhanh chóng buộc mao quản vào nhiệt kế bằng phương tiện thích hợp sao cho cột chất thử ngang với phần giữa bầu thủy ngân của nhiệt kế, điều chỉnh nhiệt kế để đầu dưới của ống mao quản cách đáy cốc 1cm. Điều chỉnh tốc độ gia nhiệt không quá 10C trong một phút. Trong lúc đó luôn khuấy nước nhẹ và quan sát cột mẫu thử, khi cột mẫu thử bắt đầu dâng lên trong ống mao quản thì đọc nhiệt độ. Kết quả đọc giữa 2 lần đo liên tiếp không được chênh lệch nhau quá 0,50C. Lấy kết quả trung bình. Nhiệt độ này được coi là điểm chảy và phải nằm trong khoảng nóng chảy quy định của các chuyên luận riêng.
XÁC ĐỊNH NHIỆT ĐỘ ĐÔNG ĐẶC: P.VL.02
Nhiệt độ đông đặc gọi tắt là điểm đông của 1 chất lỏng hoặc một chất rắn nóng chảy, là nhiệt độ cao nhất, giữ nguyên không đổi trong quá trình chuyển từ trạng thái lỏng sang trạng thái rắn.
Điểm đông của một chất lỏng chính là điểm chảy của chất đó ở trạng thái rắn, nhưng vì chất lỏng có thể chậm đông (vẫn ở trạng thái lỏng ngay ở nhiệt độ thấp hơn điểm đông) nên phải xác định điểm đông của 1 chất lỏng hoặc một chất rắn nóng chảy theo phương pháp dưới đây:
Dụng cụ:
Một ống nghiệm (a) 150mm x 25mm đặt vào trong một ống nghiệm lớn (b) khoảng 160mm x 40mm, hai ống nghiệm không chạm vào nhau và cách nhau một lớp không khí. Ống nghiệm bên trong được đậy nút có mang một que khuấy và một nhiệt kế (dài khoảng 175mm chia độ tới 0,20C), được cố định sao cho bầu nhiệt kế cách đáy ống 15mm. Que khuấy được làm bằng đũa thủy tinh hoặc bằng thép, đầu dưới uốn thành một vòng tròn nhỏ đường kính khoảng 18mm vuông góc với que khuấy. Ống nghiệm (a) và ống nghiệm (b) được giữ giữa cốc (c) có dung tích 1 lít, cốc có chứa một chất lỏng làm lạnh thích hợp. Mức chất lỏng làm lạnh cách miệng ống khoảng 20mm. Một nhiệt kế phụ trợ được giữ trong chất lỏng làm lạnh. Sơ đồ dụng cụ xác định điểm đông như sau (Hình 1):
Hình 1: Sơ đồ dụng cụ xác định điểm đông
Cách tiến hành:
Lấy lượng mẫu thử (đã được làm nóng chảy nếu cần) cho vào ống nghiệm (a) của dụng cụ sao cho bầu thủy ngân của nhiệt kế ngập trong lớp mẫu thử và xác định sơ bộ điểm đông của mẫu thử bằng cách làm lạnh nhanh. Đặt ống (a) có chứa mẫu thử vào trong bể cách thuỷ có nhiệt độ trên nhiệt độ đông đặc dự kiến khoảng 50C cho đến khi các tinh thể của mẫu thử nóng chảy hoàn toàn. Cho vào cốc (c) nước hoặc dung dịch NaCl bão hòa ở nhiệt độ thấp hơn nhiệt độ đông đặc dự kiến khoảng 50C. Lồng ống nghiệm (a) vào ống nghiệm (b) và cho vào cốc (c). Khuấy nhẹ liên tục, cứ 30 giây đọc nhiệt độ trên nhiệt kế 1 lần. Lúc đầu nhiệt độ hạ thấp dần, rồi giữ nguyên một thời gian hoặc tăng một chút, rồi giữ nguyên không đổi trong suốt thời gian đông. Để tránh hiện tượng chậm đông, khi gần đến điểm đông, có thể cho vào ống nghiệm một tinh thể nhỏ mẫu thử có sẵn hoặc cọ nhẹ vào thành trong của ống nghiệm.
XÁC ĐỊNH GÓC QUAY CỰC RIÊNG: P.VL.03
Góc quay cực của một chất là góc của mặt phẳng phân cực bị quay đi khi ánh sáng phân cực đi qua chất đó nếu là chất lỏng, hoặc qua dung dịch chất đó nếu là chất rắn.
Chất làm quay mặt phẳng phân cực theo cùng chiều kim đồng hồ được gọi là chất hữu tuyền, ký hiệu là (+). Chất làm quay mặt phẳng phân cực ngược chiều kim đồng hồ, được gọi là chất tả tuyền, ký hiệu là (-).
Nếu không có hướng dẫn riêng, góc quay cực α được xác định ở nhiệt độ 200C và với chùm tia đơn sắc có bước sóng ứng với vạch D (589,3nm) của đèn natri qua lớp chất lỏng hay dung dịch có bề mặt dày 1dm.
Góc quay cực riêng [α]D20 của một chất lỏng là góc quay cực đo được khi chùm ánh sáng D truyền qua lớp chất lỏng đó có bề dày là 1dm ở 200C, chia cho tỷ trọng của chất ở cùng nhiệt độ.
Góc quay cực riêng [α]D20 của một chất rắn là góc quay cực đo được khi chùm ánh sáng D truyền qua lớp dung dịch có bề dày là 1dm và có nồng độ là 1g/ml, ở 200C. Góc quay cực riêng của chất rắn luôn được biểu thị cùng với dung môi và nồng độ dung dịch đo. Góc quay cực riêng của một chất phải được hiểu là góc quay cực của chất này đã được sấy khô hoặc khan, trừ những trường hợp có hướng dẫn riêng. Có thể sử dụng kết quả của việc xác định sự giảm khối lượng do sấy khô hoặc hàm lượng nước trong các chuyên luận vào việc tính toán kết quả của góc quay cực riêng.
Phân cực kế
Các phân cực kế thường dùng nguồn sáng là đèn hơi natri hay hơi thủy ngân. Trong một số trường hợp, có thể cần thiết phải sử dụng phân cực kế quang điện cho phép đo lường ở các bước sóng riêng biệt. Phân cực kế phải cho phép đọc chính xác tới gần 0,010. Thang đo phải thường xuyên được kiểm định bằng các bản thạch anh chuẩn. Độ tuyến tính của thang đo phải được kiểm tra bằng dung dịch sucroza.
Cách tiến hành
Xác định góc quay cực của chất thử ở nhiệt độ 1905 đến 2005, dùng tia D của ánh sáng đèn natri phân cực, nếu không có chỉ dẫn nào khác trong chuyên luận riêng. Có thể đo ở nhiệt độ khác nếu chuyên luận riêng chỉ ra cách hiệu chỉnh nhiệt độ cho góc quay cực đo được. Tiến hành đo ít nhất 5 lần và lấy giá trị trung bình. Xác định điểm “0” của phân cực kế với ống đo rỗng khi đo chất lỏng và với ống đo chứa đầy dung môi khi đo dung dịch chất rắn.
Tính góc quay cực riêng theo các biểu thức sau:
Cho chất lỏng
[α]D20 =
Cho chất rắn
[α]D20 = x 100
Trong đó:
α: góc quay cực đo được
l: chiều dài ống đo của phân cực kế, đơn vị: dm.
d: tỷ trọng của chất thử (lỏng)
c: nồng độ % của chất thử (rắn) trong dung dịch.
HƯỚNG DẪN PHƯƠNG PHÁP XÁC ĐỊNH CÁC THÀNH PHẦN VÔ CƠ: P.VC
XÁC ĐỊNH GIẢM KHỐI LƯỢNG KHI LÀM KHÔ: P.VC.01
Tiến hành:
Nếu không có chỉ dẫn gì khác trong các chuyên luận riêng, trộn đều mẫu và cân chính xác khoảng 1 - 2g mẫu. Nếu mẫu là tinh thể thì nghiền nhỏ.
Cho mẫu vào cốc chuyên dụng kiểm tra độ ẩm (cốc có nắp thuỷ tinh, đã được làm khô trong 30 phút với các điều kiện áp dụng để phân tích độ ẩm trong mẫu, đã cân bì trước), đậy nắp, cân mẫu và cốc.
Dàn đều mẫu trong đáy cốc sao cho lớp mẫu không dày quá 5mm, trong trường hợp mẫu dày, xốp thì chiều dày không quá 10mm. Đặt cốc chứa mẫu vào buồng làm khô, mở nắp và để nắp trong buồng làm khô. Làm khô mẫu tại nhiệt độ và thời gian đã được chỉ ra trong từng chuyên luận quy định riêng. Ngay khi mở buồng làm khô đậy ngay nắp cốc, để cốc nguội tới nhiệt độ phòng trong bình hút ẩm. Cân cốc chứa mẫu đã làm khô, tính khối lượng mẫu đã làm khô.
Nếu điểm nóng chảy của mẫu nhỏ hơn nhiệt độ được quy định cho việc thử sự giảm khối lượng khi làm khô. Chuẩn bị mẫu như trên, sau đó đặt cốc chứa mẫu trong bình hút ẩm chứa H2SO4 đặc, đậy nắp bình hút ẩm và hút duy trì áp suất trong bình tại 130Pa (1mmHg), làm khô mẫu tại điều kiện này trong 24giờ. Cân khối lượng bì và mẫu, tính khối lượng mẫu đã làm khô.
XÁC ĐỊNH GIẢM KHỐI LƯỢNG KHI NUNG: P.VC.02
Tiến hành:
Nếu không có chỉ dẫn gì khác trong các chuyên luận riêng, tiến hành chuẩn bị như mô tả trong phương pháp “Xác định giảm khối lượng khi làm khô - P.VC.01”, sử dụng chén nung bằng Platin hoặc thạch anh. Nung mẫu tại nhiệt độ 4500C - 5000C.
XÁC ĐỊNH HÀM LƯỢNG NƯỚC: P.VC.03
Phương pháp Karl - Fischer
Nguyên tắc
Phương pháp định lượng nước này dựa trên phản ứng toàn lượng của nước với lưu huỳnh dioxyd và iod trong dung môi khan chứa một chất bazơ hữu cơ thích hợp. Dung môi hữu cơ thông dụng là metanol khan nước, cũng có khi được thay bằng dung môi hữu cơ khác thích hợp để hòa tan chất thử. Chất bazơ hữu cơ là pyridin, nhưng hiện nay người ta đã dùng những chất bazơ hữu cơ khác để thay thế như imidazol, 2-methylaminopyridin.
Dụng cụ
Hiện nay có nhiều loại dụng cụ, nhưng nguyên tắc đều phải cấu tạo sao cho thao tác thuận tiện và tránh ẩm. Dụng cụ gồm các loại cốc chuẩn độ dung tích khoảng 60ml, có nắp gắn điện cực platin kép, một ống dẫn khí nitơ, có lỗ cắm với buret và lỗ cắm ống thông hơi chứa chất hút ẩm, có gắn máy khuấy điện hoặc khuấy từ. Chất thử cho vào bình chuẩn độ qua lỗ trên nắp hoặc miệng bên cạnh có nút mài. Điểm kết thúc phản ứng được xác định bằng điện kế gắn trong mạch có biến trở 2000Ω, nối với một nguồn pin 1,5 vôn. Lúc bắt đầu kim điện kế chỉ điểm không, vì dòng diện lúc chạy qua 2 điện cực platin không đáng kể. Khi nhỏ thuốc thử Karl-Fischer vào dung dịch, do hiện tượng khử cực nên kim điện kế lệch đi nhưng lập tức trở về vị trí ban đầu, chỉ khi đến điểm tương đương thì một giọt thuốc thử thừa sẽ làm kim lệch đi và duy trì ít nhất 30 giây.
Thuốc thử
Thuốc thử Karl-Fischer gốc gồm 4 thành phần chính là lưu huỳnh dioxyd, iod, pyridin hoặc một bazơ hữu cơ khác và metanol pha thành một dung dịch, hoặc hai dung dịch. Trường hợp pha thành hai dung dịch thì dung dịch A chứa lưu huỳnh dioxyd và pyridin pha trong metanol khan, dung dịch B chứa iod pha trong metanol khan. Trước khi dùng một giờ, trộn đều 1 thể tích dung dịch A với 1 thể tích dung dịch B, sau đó xác định đương lượng nước của thuốc thử. Lượng thuốc thử thu được chỉ dùng trong ngày. Hiện nay trên thị trường vừa có các loại thuốc thử như trên. vừa có loại đã thay pyridin và metanol bằng chất bazơ hữu cơ khác và dung môi hữu cơ khác. Do vậy, cần kiểm tra kỹ thành phần thuốc thử và cách sử dụng cho đúng mục đích. Các thuốc thử Karl-Fischer và dung môi dùng trong phương pháp đều phải khan nước, bảo quản trong lọ màu, tránh ánh sáng, chống ẩm và phải xác định lại đương lượng nước trước khi dùng.
Xác định đương lượng của thuốc thử
Đương lượng của thuốc thử Karl-Fischer dễ bị thay đổi theo thời gian nên trước khi dùng phải xác định lại và phải đạt tối thiểu 3,5mg nước cho 1ml thuốc thử. Có thể xác định theo 2 cách:
a. Dùng một hóa chất có hàm lượng nước kết tinh xác định, sau khi đã sấy ở nhiệt độ quy định đến khối lượng không đổi để loại hết ẩm, cho tác dụng với thuốc thử rồi tính ra đương lượng. Thường dùng natri tartrat dihydrat (C4H2Na2.2H2O)
Cách tiến hành như sau:
Cho một lượng metanol hoặc dung môi thích hợp dùng cho thuốc thử Karl-Fischer vào cốc chuẩn độ đủ ngập điện cực platin rồi nhỏ thuốc thử Karl-Fischer đến điểm tương đương (kim điện kế lệch đi tối thiểu 30 giây). Cho nhanh khoảng từ 250mg đến 350mg natri tartrat dihydrat đã cân chính xác vào cốc và chuẩn độ bằng thuốc thử Karl-Fischer đến điểm dừng, tính đương lượng (F) của thuốc thử theo công thức:
F = 2.(18,02/230,08).(W/V)
Trong đó:
18,02 và 230,08: khối lượng phân tử của nước và của natri tartrat dihydrat
W: khối lượng natri tartrat dihydrat tính bằng mg.
V: thể tích của thuốc thử đã dùng (tính bằng ml).
Trường hợp này áp dụng cho việc xác định hàm lượng nước nhỏ dưới 1%.
b. Trường hợp để xác định một lượng nước lớn hơn 1%, người ta dùng nước tinh khiết đã chưng cất đạt tiêu chuẩn làm chất chuẩn hòa vào metanol hoặc dung môi thích hợp loại dùng cho thuốc thử Karl-Fischer rồi dùng thuốc thử Karl-Fischer để chuẩn độ.
Cách làm như sau: Cho 25ml metanol vào cốc chuẩn độ, chuẩn độ bằng thuốc thử Karl-Fischer đến điểm dừng. Thêm nhanh khoảng 50mg nước tinh khiết đã cân chính xác vào cốc chuẩn độ trên và chuẩn độ bằng thuốc thử Karl-Fischer đến điểm tương đương. Tính đương lượng nước F theo công thức:
F = W/V
Trong đó:
W: khối lượng nước đã cho vào tính bằng mg
V: thể tích thuốc thử đã dùng tính bằng ml
Tiến hành định lượng
Chuẩn bị mẫu thử:
Nếu không có chỉ dẫn gì khác trong chuyên luận, người ta cân hoặc lấy chính xác một lượng mẫu ước lượng chứa khoảng 10mg đến 50mg nước đem định lượng. Thao tác phải nhanh và thực hiện trong phòng có độ ẩm thấp để tránh ẩm ở ngoài ảnh hưởng đến chất phân tích.
Phương pháp định lượng trực tiếp
Nếu không có chỉ dẫn gì khác trong chuyên luận, người ta cho khoảng 20ml metanol khan vào cốc chuẩn độ, nhỏ thuốc thử Karl-Fischer đến điểm tương đương, cho nhanh lượng chất thử đã cân vào cốc chuẩn độ, đóng nút ngay, khuấy đều để phản ứng tác dụng trong khoảng 1 phút rồi tiếp tục chuẩn độ bằng thuốc thử Karl-Fischer đến điểm tương đương, ghi thể tích (ml) thuốc thử đã dùng sau khi đã cho chất thử (V). Tính kết quả theo công thức:
X (khối lượng nước có trong lượng mẫu thử - đơn vị tính: mg) = V.F
Trong đó:
V: số ml thuốc thử đã dùng cho lần chuẩn độ sau khi cho chất thử
F: hệ số đương lượng nước của thuốc thử Karl-Fischer
Phương pháp định lượng gián tiếp
Pha dung dịch nước chuẩn: cho 500ml metanol hoặc dung môi thích hợp vào bình định mức 1 lít, lấy chính xác 2,0ml nước tinh khiết cho vào bình trên và bổ sung dung môi đủ 1000ml. Lấy chính xác 25,0ml dung dịch này cho vào cốc định lượng và chuẩn độ bằng thuốc thử Karl-Fischer vừa mới xác định độ chuẩn. Ghi thể tích (V-ml) thuốc thử Karl-Fischer đã dùng. Tính hàm lượng nước của dung dịch nước chuẩn theo công thức:
W (mg/ml) = V.F/25
Trong đó:
V: số ml thuốc thử Karl-Fischer đã dùng.
F: hệ số đương lượng nước của thuốc thử Karl-Fischer
Tiến hành: Nếu không có chỉ dẫn gì khác, người ta cho một lượng metanol, hoặc dung môi thích hợp khan vào cốc định lượng vừa đủ ngập điện cực, chuẩn độ bằng thuốc thử Karl-Fischer đến điểm tương đương. Cho nhanh lượng chất thử đá cân vào, đóng nút ngay, cho tiếp một lượng thuốc thử Karl-Fischer chính xác vào cốc sao cho thừa khoảng 1ml, hoặc theo một lượng đã quy định trong chuyên luận riêng. Đóng nút, để yên 1 phút, tránh ánh sáng, thỉnh thoảng khuấy. Chuẩn độ phần thuốc thử Karl-Fischer thừa bằng dung dịch nước chuẩn vừa mới pha trên. Tính hàm lượng nước (mg) có trong mẫu thử theo công thức:
A (mg) = F.V1 - W.V2
Trong đó:
F: đương lượng nước của thuốc thử Karl-Fischer
V1: số ml thuốc thử Karl-Fischer đã cho vào mẫu thử
V2: số ml dung dịch nước chuẩn đã dùng
W: hàm lượng nước tính bằng mg/ml của dung dịch nước chuẩn ở trên.
Chú ý:
Cần phải kiểm tra chất thử có tương kỵ với thuốc thử Karl-Fischer hay không trước khi áp dụng phương pháp này. Những chất có khả năng phản ứng với một hay nhiều thành phần của thuốc thử như acid ascorbic, các mercaptan, các sulfid, các muối hydrocarbonat và carbonat kiềm, các oxyd và hydrat của oxyd kim loại… không áp dụng được phương pháp này. Đối với các aldehyd và ceton, hiện nay đã có loại thuốc thử dành riêng để định lượng nước trong các chất này.
Những dung môi hữu cơ sau đây có thể dùng thay thế metanol trong thuốc thử Karl-Fischer khi chất thử không tan trong metanol: cloroform, metyl celosolve, dietylen glycol monoetyl ete. Trước khi sử dụng phải làm khan bằng zeolit đạt tiêu chuẩn cho định lượng nước.
Những chất bazơ hữu cơ sau đây có thể thay pyridin trong thuốc thử Karl-Fischer: imidazol, 2-metylaminopyridin. Phải kiểm tra hàm lượng nước trước khi dùng.
XÁC ĐỊNH HÀM LƯỢNG CLORID: P.VC.04
Chuẩn bị mẫu thử:
Nếu không có chỉ dẫn gì khác, lấy mẫu thử (khối lượng cụ thể theo chỉ dẫn trong các chuyên luận riêng) vào trong ống nghiệm nessler, hòa tan với khoảng 30ml nước cất, thử pH dung dịch, nếu kiềm tính, trung hòa bằng dung dịch acid nitric loãng TT.170.
Thêm 6ml dung dịch acid nitric loãng TT.170 và pha loãng với nước cất tới 50ml. Nếu mẫu là chất lỏng, lấy mẫu vào ống nghiệm nessler (thể tích cụ thể theo chỉ dẫn trong các chuyên luận riêng), pha loãng đến 50ml bằng nước cất (ống A).
Chuẩn bị dung dịch chuẩn:
Lấy dung dịch chuẩn HCl 0,01N (thể tích cụ thể quy định trong các chuyên luận riêng) vào ống nghiệm nessler, thêm 6ml dung dịch acid nitric loãng TT.170, thêm nước cất đến 50ml (ống B).
Nếu ống A không trong, tiến hành lọc cả hai ống A và B trong cùng điều kiện. Thêm 1ml dung dịch bạc nitrat TT.215 vào cả 2 ống, lắc đều hỗn hợp, để yên trong 5 phút, tránh ánh sáng chiếu trực tiếp.
So sánh độ đục của hai dung dịch:
Quan sát cả hai ống từ trên xuống (trên nền là một mặt phẳng màu đen). Độ đục của ống A không được vượt quá độ đục của ống B.
XÁC ĐỊNH HÀM LƯỢNG SULFAT: P.VC.05
Chuẩn bị dung dịch thử:
Nếu không có chỉ dẫn gì khác, cân mẫu thử (khối lượng cụ thể theo chỉ dẫn trong các chuyên luận riêng) cho vào trong ống nghiệm nessler, hòa tan với khoảng 30ml nước cất, thử pH dung dịch, nếu kiềm tiến hành trung hòa bằng dung dịch acid hydrocloric loãng TT.127. Thêm 1ml dung dịch acid hydrocloric loãng TT.127 và pha loãng với nước cất tới 50ml (ống A).
Nếu mẫu là chất lỏng lấy mẫu (thể tích cụ thể theo chỉ dẫn trong các chuyên luận riêng) vào ống nghiệm nessler, pha loãng đến 50ml bằng nước cất (ống A).
Chuẩn bị dung dịch chuẩn:
Lấy dung dịch chuẩn H2SO4 0,01N vào ống nghiệm nessler (Thể tích cụ thể quy định trong các chuyên luận riêng), thêm 1ml dung dịch acid hydrocloric loãng TT.127, thêm nước cất đến 50ml (ống B).
Nếu ống A không trong, tiến hành lọc cả hai ống A và B trong cùng điều kiện. Thêm 2ml dung dịch bari clorid TT.035 vào cả 2 ống, lắc đều hỗn hợp, để yên trong 10 phút.
So sánh độ đục của hai dung dịch:
Quan sát cả 2 ống nghiệm từ trên xuống (trên nền là một mặt phẳng màu đen). Độ đục của ống A không được vượt quá độ đục của ống B.
XÁC ĐỊNH HÀM LƯỢNG FLO: P.VC.06
Phương pháp so màu với thuốc thử tạo màu là Th(NO3)4
Chú ý:
Khi áp dụng phương pháp này đối với các hợp chất hữu cơ, nhiệt độ của quá trình chưng cất là 1350C đến 1400C và phải được kiểm tra nghiêm ngặt thường xuyên tránh khả năng gây nổ
Để giảm thiểu ảnh hưởng của flo thôi ra từ thiết bị chưng cất, thiết bị chưng cất sẽ được xử lý trước như sau:
Xử lý đồ thuỷ tinh với dung dịch NaOH 10% nóng, tiếp theo xả dưới vòi nước và rửa lại bằng nước cất. Ít nhất một lần trong một ngày, đun sôi với 15 - 20ml dung dịch H2SO4 loãng (~50%) cho đến khi có khói thoát ra, để nguội, trút hết acid và xử lý lại lần nữa với NaOH 10% và tráng lại bằng nước cất.
Nên sử dụng các bình và dụng cụ bằng Teflon khi xác định Flo.
Tiến hành:
Nếu không có chỉ dẫn gì khác. Lấy 5,0g mẫu và 30ml nước cất vào trong bình chưng cất có ống nhánh và khóa, ống này được nối với sinh hàn, có gắn nhiệt kế và ống mao quản. Một đầu nhiệt kế và ống mao quản nhúng vào chất lỏng.
Thêm cẩn thận, vừa thêm vừa khuấy 10ml HClO4 đặc, thêm 2 hoặc 3 giọt dung dịch AgNO3 (1 trong 2) và vài viên bi thuỷ tinh. Nối ống đun với phễu nhỏ giọt nhỏ hoặc với bộ phận sinh hơi nước qua ống mao quản. Đỡ ống bằng miếng lót amiăng có lỗ sao cho 1/3 thân ống có thể tiếp xúc với nguồn nhiệt. Chưng cất cho đến khi nhiệt độ đạt 1350C. Thêm nước vào từ phễu hoặc sục hơi nước qua ống mao quản, duy trì nhiệt độ từ 1350C đến 1400C trong suốt quá trình này. Tiếp tục chưng cất cho đến khi thu được 100ml dịch chưng cất. Thu 100ml dịch chưng cất đầu tiên (dung dịch A). Sau đó thu tiếp 50ml dịch chưng nữa (dung dịch B) để chắc chắn flo trong mẫu đã được hóa hơi hoàn toàn.
Lấy 50ml dung dịch A cho vào trong ống nghiệm nessler, trong ống nghiệm nessler khác cho 50ml nước cất đã được chưng cất qua thiết bị trên, ống này được coi như là mẫu so sánh. Thêm vào mỗi ống 0,1ml dung dịch natri alizarin sunfonat 0,1% đã lọc trước và 1ml dung dịch hydroxylamin hydrocloric 0,025%, trộn đều hỗn hợp. Thêm từng giọt, vừa thêm vừa khuấy một lượng dung dịch NaOH 1N hoặc 0,05N vào ống chứa dung dịch A cho đến khi ống chứa dung dịch A có màu hồng nhạt giống với màu dung dịch so sánh. Tiếp theo, thêm vào mỗi ống 1,0ml dung dịch HCl 0,1N và khuấy kỹ. Dùng buret có chia độ (0,05m/vạch) thêm từ từ (vừa thêm vừa khuấy đều) vào ống chứa chưng cất dung dịch Th(NO3)4 0,025%, đến khi dung dịch trong ống có màu nhạt. Ghi lại thể tích của dung dịch Th(NO3)4 đã cho vào ống chứa dịch chưng cất, tiếp theo thêm chính xác lượng tương tự vào ống chứa dung dịch so sánh, khuấy đều hỗn hợp. Dùng buret thêm vào dung dịch so sánh dung dịch NaF (10μF-/ml) TT.228 tới khi màu của hai ống giống nhau sau khi pha loãng tới thể tích như nhau. Khuấy kỹ hỗn hợp, đuổi hết các bọt khí ra khỏi dung dịch trước khi so sánh màu. Kiểm tra điểm tương đương bằng cách thêm 1 hoặc 2 giọt dung dịch NaF TT.228 vào mẫu thử. Sự chuyển màu rõ ràng sẽ xảy ra. Ghi thể tích của dung dịch NaF TT.228 thêm vào.
Pha loãng dung dịch B tới 100ml và khuấy hỗn hợp thật kỹ. Lấy 50ml dung dịch B pha loãng này cho vào ống nghiệm nessler và tiến hành thực hiện như tiến hành với dung dịch A.
Đánh giá kết quả:
Tổng thể tích của dung dịch NaF sử dụng khi chuẩn độ dung dịch A và dung dịch B là không được vượt quá 2,5ml.
XÁC ĐỊNH HÀM LƯỢNG CHÌ: P.VC.07
Nguyên tắc:
Dithizon tạo phức màu đỏ với chì. Chì dithizonat được chiết chọn lọc và loại trừ các nguyên tố ảnh hưởng bằng kali cyanid. Chiết phức màu đỏ bằng dung dịch Dithizon pha trong cloroform rồi đo độ hấp thụ quang của dung dịch chiết.
Hóa chất và thuốc thử:
- Dung dịch Amoni-cyanid: hoà tan 2g KCN trong 15ml dung dịch amoniac đặc TT.010, pha loãng với nước cất tới 100ml.
- Acid hydrocloric loãng: nồng độ khoảng 10% (m/v)
- Dung dịch Pb chuẩn gốc chứa 1000μg Pb/1ml
- Dung dịch Pb chuẩn pha loãng (10μg Pb trong 1ml): Pha ngay trước khi dùng. Lấy 1ml dung dịch Pb chuẩn gốc vào bình định mức 100ml, định mức tới vạch bằng HNO3 1%.
Thiết bị và dụng cụ:
- Cân phân tích có độ chính xác 0,1mg
- Bếp điện
- Pipet, bình định mức, cốc đong, que khuấy, phễu chiết…
- Bi thủy tinh
Chú ý:
Các dụng cụ thí nghiệm phải được làm sạch trước khi tiến hành thí nghiệm và tráng bằng HNO3 loãng nóng.
Chuẩn bị dung dịch thử:
Chú ý:
Khi tiến hành vô cơ hóa một số hợp chất hữu cơ bằng H2O2 có thể gây nổ, tất cả các cảnh báo an toàn kỹ thuật cần được áp dụng trong suốt quá trình thí nghiệm. Quá trình vô cơ hóa mẫu phải được tiến hành hoàn toàn trong tủ hút.
Nếu có chỉ dẫn trong các chuyên luận riêng, chuẩn bị dung dịch thử theo các chỉ dẫn trong chuyên luận riêng. Trong trường hợp không có chỉ dẫn riêng, đối với mẫu thử là các hợp chất hữu cơ, dung dịch thử được chuẩn bị như sau:
Cân chính xác 1g mẫu thử, cho vào bình vô cơ hóa, thêm 5ml H2SO4 đặc và vài viên bi thủy tinh. Tiến hành vô cơ hóa tại nhiệt độ không quá 1200C trên bếp điện đến khi bắt đầu quá trình than hóa (lượng H2SO4 thêm vào phải tẩm ướt hoàn toàn mẫu nhưng không quá 10ml).
Sau khi mẫu bắt đầu bị acid phá hủy thêm cẩn thận từng giọt H2O2 30%, thêm từ từ từng giọt để kiểm soát tốc độ phản ứng, nếu tốc độ phản ứng xảy ra nhanh có thể giảm nhiệt độ đun, ngừng đun nóng nếu quá nhiều bọt. Khuấy đều dung dịch trong bình để tránh hiện tượng các chất bám dính vào thành bình không phản ứng. Khi dung dịch bắt đầu thẫm màu dần, thêm lượng nhỏ H2O2, tiếp tục vô cơ hóa đến khi các thành phần hữu cơ bị phá hủy, tăng nhiệt độ đun lên 2500C - 3000C đến khi khói trắng SO3 bay ra và dung dịch trở nên trong không màu hoặc màu vàng nhạt. Để nguội, thêm cẩn thận 10ml nước cất, tiếp tục đun đến khi có khói thoát ra nhiều, để nguội. Định mức bằng nước cất đến thể tích phù hợp thu được dung dịch thử.
Tiến hành:
Nếu không có chỉ dẫn gì khác trong các chuyên luận riêng, chuyển dung dịch thử vào phễu chiết. Thêm 6ml dung dịch amoni citrat (PbT) TT.011, 2ml dung dịch hydroxylamin hydroclorid TT.132 (khi phân tích Pb trong các muối Fe dùng 10ml dung dịch amoni citrat (PbT) TT.011) và 2 giọt dung dịch phenol đỏ TT.178. Kiềm hóa dung dịch bằng cách thêm dung dịch amoniac đặc TT.010 (dung dịch bắt đầu xuất hiện màu hồng). Làm nguội dung dịch dưới vòi nước chảy nếu cần, thêm tiếp 2ml dung dịch kali cyanid (PbT) TT.193. Tiến hành chiết ngay dung dịch với 5ml dung dịch dithizon chiết TT.096, chiết dung dịch dithizon ra những phễu chiết riêng biệt, tiến hành như vậy cho đến khi dung dịch Dithizon chiết ra có màu xanh. Gộp các dịch chiết Dithizon và lắc trong 30 giây với 20ml dung dịch HNO3 loãng 1%, loại bỏ lớp cloroform, thêm vào phần dung dịch acid còn lại 5ml dung dịch Dithizon chuẩn TT.097 và 4ml dung dịch Amoni-cyanid, lắc tiếp trong 30 giây. Dung dịch Dithizon này có màu đỏ.
Tiến hành tương tự đối với dung dịch chuẩn Pb chứa Pb2+, thể tích dung dịch Pb chuẩn theo chỉ dẫn trong các chuyên luận riêng.
Đánh giá kết quả:
Màu đỏ tía của dung dịch cloroform trong thử nghiệm đối với dung dịch thử (phức chì Dithizonat) không được đậm hơn màu dung dịch cloroform trong thử nghiệm đối với dung dịch Pb chuẩn.
XÁC ĐỊNH HÀM LƯỢNG ASEN: P.VC.08
Nguyên tắc:
As3+ tạo phức màu với bạc dietyldithiocacbamat. Xác định hàm lượng As dựa trên độ hấp thụ quang của phức chất đó tại bước sóng 535 đến 540nm.
Hóa chất - thuốc thử: Tất cả các hóa chất thuốc thử sử dụng trong quy trình phân tích này phải tinh khiết và có hàm lượng As thấp (nhỏ hơn giới hạn phát hiện).
- Dung dịch bạc dietyldithiocacbamat
Hòa tan 1g tinh thể bạc dietyldithiocacbamat (C2H5)2NCSSAg trong 200ml pyridin (thực hiện trong tủ hút). Giữ dung dịch này trong lọ thủy tinh màu, tránh ánh sáng và sử dụng trong 1 tháng.
- Bạc dietyldithiocacbamat sẵn có trên thị trường hoặc có thể được chuẩn bị như sau:
+ Hòa tan 1,7g bạc nitrat với 100ml nước cất trong cốc thủy tinh.
+ Hoà tan 2,3g natri dietyldithiocacbamat trong 100ml nước trong cốc khác và lọc.
+ Cả hai dung dịch để nguội tới khoảng 150C, trộn hỗn hợp hai dung dịch với nhau khuấy trộn kỹ.
+ Lọc lấy kết tủa, rửa với khoảng 200ml nước lạnh.
- Kết tinh lại thuốc thử Bạc dietyldithiocacbamat:
+ Hoà tan 1g thuốc thử Bạc dietyldithiocacbamat trong 100ml dung môi pyridin vừa được chưng cất rồi lọc hỗn hợp.
+ Thêm một thể tích tương ứng nước lạnh vào dung dịch pyridin và khuấy trộn đều.
+ Lọc kết tủa và rửa kết tủa bằng nước lạnh rồi sấy khô trong chân không ở nhiệt độ phòng từ 2 đến 3 giờ.
+ Bạc dietyldithiocacbamat là tinh thể tinh khiết màu vàng, không bị biến đổi trong vòng 1 tháng khi bảo quản trong lọ tránh ánh sáng. Loại bỏ khi đã biến màu hoặc có mùi.
- Dung dịch chuẩn gốc As: Dung dịch nồng độ 1000mg/l.
- Dung dịch chuẩn làm việc As nồng độ 1μg/ml: lấy 1ml dung dịch As gốc cho vào bình định mức 1000ml, thêm 10ml dung dịch acid sulfuric loãng TT.246 rồi định mức với nước cất đến 1000ml. Dung dịch này dùng trong 3 ngày.
- Dung dịch SnCl2: Hoà tan 40g SnCl2.2H2O vào trong 100ml dung dịch HCl 6M. Bảo quản dung dịch trong lọ thủy tinh và dùng trong 3 tháng.
- Bông tẩm Pb(CH3COO)2. Ngâm bông trong dung dịch bão hòa Pb(CH3COO)2, vắt hết dung dịch thừa và làm khô trong chân không ở nhiệt độ phòng. Chú ý khi chuẩn bị và sử dụng bông phải cẩn thận tránh bị ô nhiễm chì.
Thiết bị:
- Cân phân tích có độ chính xác 0,1mg
- Dụng cụ Asin hóa (hình 2).
Hình 2: Sơ đồ dụng cụ asin hóa
Chuẩn bị mẫu:
Dung dịch mẫu của các hợp chất hữu cơ được chuẩn bị trong bình A theo chỉ dẫn cụ thể trong chuyên luận riêng.
Chú ý:
Phải đeo kính bảo hộ và đeo găng tay khi sử dụng oxy già.
Nếu trong mẫu có hợp chất chứa halogen, để tránh mất As3+ đun mẫu với H2SO4 ở nhiệt độ thấp, không đun sôi hỗn hợp và thêm cẩn thận oxy già trước khi mẫu bắt đầu hóa than.
Nếu không có chỉ dẫn nào khác, cân 1g mẫu, chuyển vào bình A, thêm 5ml H2SO4 đặc và một vài hạt bi thủy tinh, phân hủy mẫu ở nhiệt độ ≤1200C trên bếp điện đặt trong tủ hốt (thêm H2SO4 nếu cần thiết để làm ướt mẫu nhưng tổng thể tích thêm không quá 10ml). Sau khi mẫu đã bắt đầu phân hủy bởi acid, thêm cẩn thận từng giọt oxy già 30%, thêm từ từ từng giọt oxy già để kiểm soát tốc độ phản ứng nếu tốc độ phản ứng xảy ra nhanh có thể giảm nhiệt độ đun, ngừng đun nóng nếu quá nhiều bọt. Khuấy trộn dung dịch trong bình để tránh hiện tượng các chất bám dính vào thành bình không phản ứng. Tiếp tục phân hủy mẫu cho đến khi các chất hữu cơ trong mẫu bị phá hủy, từ từ nâng nhiệt độ của bếp điện lên đến 250 - 3000C cho tới khi khói của H2SO4 bay ra và dung dịch trở lên không màu hoặc có vàng rơm nhạt, sau đó làm nguội. Thêm 10ml nước cất và lại làm bay hơi (khói của H2SO4 bay ra) rồi để nguội. Thêm cẩn thận 10ml nước để rửa thành bình và pha loãng mẫu tới 35ml (dịch thử).
Có thể không tiến hành chuẩn bị dung dịch thử trong bình A (bình sinh asin), trong trường hợp như vậy, sau khi đã chuẩn bị xong dung dịch thử, lấy thể tích dung dịch thử tương đương với 1g mẫu vào bình A, thêm nước cất đến đủ 35ml.
Tiến hành:
Lấy bình A đã chứa 35ml dung dịch thử.
Thêm 20ml dung dịch H2SO4 loãng (khoảng 20%), 2ml dung dịch KI TT.200 và 0,5ml dung dịch SnCl2, khuấy đều. Để yên hỗn hợp phản ứng trong 30 phút ở nhiệt độ phòng. Bộ ống lọc khí C của thiết bị được nhét hai nút bông tẩm Pb(CH3COO)2, hai nút bông được đặt cách nhau một khoảng trong ống, bôi mỡ tại các đầu nối B và D nếu cần thiết và nối hệ thống lọc khí với ống hấp thụ E (xem hình vẽ). Chuyển 3ml dung dịch bạc dietyldithiocacbamat vào ống hấp thụ, thêm 3g Zn hạt vào hỗn hợp trong bình sinh khí (bình A), lắp ngay cổ bình A với đầu nối B của ống lọc khí.
Để các phản ứng (phản ứng tạo hydro mới sinh, phản ứng tạo màu) xảy ra ở nhiệt độ phòng (250C ± 30C) trong 45 phút, khuấy dung dịch phản ứng 10 phút/lần. (Thêm lượng nhỏ iso propanol vào bình A để ổn định được tốc độ sinh khí). Tháo ống hấp phụ ra khỏi bình A và hệ thống lọc khí, chuyển dung dịch bạc dietyldithiocacbamat trong ống E vào cuvet đo quang. Xác định độ hấp thụ quang của phức tại bước sóng hấp thụ cực đại từ 535nm đến 540nm, sử dụng dung dịch bạc dietyldithiocacbamat làm mẫu trắng.
Tiến hành tương tự với 3ml dung dịch chuẩn As 1μg/ml (dung dịch chuẩn). Nhiệt độ của quá trình sinh asin trong thử nghiệm của dung dịch chuẩn và thử nghiệm của dung dịch thử phải ổn định, không được lệch nhau quá 20C.
Đánh giá kết quả:
Độ hấp thụ quang của phức chất tạo bởi As trong dung dịch thử với dung dịch bạc dietyldithiocacbamat không được vượt quá độ hấp thụ quang phức chất tạo bởi dung dịch chuẩn (chứa 3μg As) với dung dịch bạc dietyldithiocacbamat.
Chú ý:
Các kim loại hoặc các muối của các kim loại như Cr, Co, Cu, Hg, Mo, Ni, Pd, Ag có thể gây nhiễu cho quá trình sinh asin. Sb - kim loại có thể sinh stibin là kim loại duy nhất có thể gây nhiễu dương tính cho quá trình tạo màu với thuốc thử bạc dietyldithiocacbamat. Stibin tạo phức màu đỏ có cực đại hấp thụ tại bước sóng 510nm nhưng tại bước sóng từ 535nm đến 540nm độ hấp thụ quang của phức Sb-DDC (Sb-dietyldithiocacbamat) giảm thiểu đáng kể và không còn gây ảnh hưởng tới kết quả phân tích.
XÁC ĐỊNH HÀM LƯỢNG MỘT SỐ KIM LOẠI NẶNG BẰNG PHƯƠNG PHÁP QUANG PHỔ HẤP THỤ NGUYÊN TỬ (AAS): P.VC.09
Nguyên tắc:
Mẫu được hoà tan trong acid, vô cơ hóa bằng hỗn hợp H2SO4 và HNO3, trong một vài trường hợp có HClO4. Sử dụng phương pháp Quang phổ hấp thụ nguyên tử kỹ thuật ngọn lửa để phân tích các kim loại Cd, Pb, Cu, Cr, Zn. Kim loại As được phân tích bằng kỹ thuật hydrid hóa.
Chú ý chung:
Phân tích vi lượng kim loại yêu cầu đặc biệt chú trọng các điều kiện chuẩn bị thí nghiệm nhằm giảm thiểu sự ảnh hưởng của các kim loại có trong hóa chất, dụng cụ tới quá trình phân tích càng nhỏ càng tốt. Trước khi sử dụng, tất cả các dụng cụ thủy tinh cần được làm sạch trước bằng dung dịch acid loãng nóng (1 phần HCl, 1 phần HNO3 đặc và 3 phần nước cất), sau đó rửa sạch bằng nước cất. Các hóa chất, thuốc thử sử dụng trong quá trình phân tích phải là loại đặc biệt tinh khiết (có hàm lượng kim loại thấp) sử dụng cho phân tích vết kim loại.
Dụng cụ, thiết bị:
Bình Kjeldahl phá mẫu bằng thủy tinh thạch anh hoặc thủy tinh borosilicat (thông thường có dung tích 100ml). Bộ dụng cụ phá mẫu như hình 3:
Hình 3: Bộ Kjeldahl phá mẫu
Máy Quang phổ hấp thụ nguyên tử: Tất cả các máy quang phổ hấp thụ nguyên tử (AAS) có thể sử dụng tuỳ chọn nhiên liệu cháy: hỗn hợp giữa không khí với N2O, H2, C2H2 và có thể tiến hành đo tại dải bước sóng bức xạ từ 180 - 600nm.
Hóa chất, chất chuẩn:
- HNO3 đặc (65%, d = 1,42)
- Dung dịch HClO4 60% (m/m)
- H2SO4 98%
- HCl đặc (36%, d = 1,16)
- Dung dịch HCl 5N.
- Na2SO4 (PA)
- KCl (PA)
- Nước cất, không có kim loại (nước cất hai lần và làm tinh khiết qua cột trao đổi cation, VD cột Amberlite IR 120 H)
• Chất chuẩn gốc Cu, Zn, Cr, Sb, Pb, Cd 1000mg/l.
Pha dung dịch chuẩn trung gian: Cu, Zn, Cr, Sb (100μg/ml); Pb, Cd (20μg/ml)
Pha dung dịch chuẩn làm việc:
Lấy vào các bình định mức 100ml lần lượt 0; 1; 2; 3; 4; 5ml dung dịch chuẩn thích hợp. Pha loãng tới khoảng 50ml, thêm 2ml H2SO4 đặc và 10ml HCl đặc, lắc đều hỗn hợp. Định mức tới vạch bằng nước cất.
Dung dịch này có nồng độ 0; 0,2; 0,4; 0,6; 0,8; và 1,0μg/ml đối với Cd và Pb. Nồng độ 0; 1,0; 2,0; 3,0; 4,0; và 5,0μg/ml đối với Cu, Zn, Cr và Sb.
Chuẩn bị dung dịch thử:
Phương pháp 1 (sử dụng cho các hợp chất tan được trong dung dịch acid loãng):
Cân chính xác khoảng 2,5g mẫu, hoà tan trong hỗn hợp acid (4ml H2SO4 đặc và 5ml HCl đặc). Chuyển dung dịch này vào bình định mức 50ml, nếu phân tích Ba có thể lấy dung dịch này làm dung dịch thử. Trong trường hợp phân tích các kim loại nặng khác thêm 0,0954g KCl. Định mức đến vạch bằng nước cất. Dịch này là dung dịch thử (dung dịch A).
Phương pháp 2 (sử dụng cho các hợp chất không tan trong dung dịch acid loãng):
Cân chính xác khoảng 2,5g mẫu vào trong bình Kjeldahl dung tích 100ml ~ 150ml, thêm 5ml HNO3 loãng. Ngay khi phản ứng ban đầu giảm xuống, đun nóng nhẹ cho đến khi phản ứng xảy ra mạnh hơn rồi ngừng lại, để nguội. Thêm 4ml H2SO4 đặc với tốc độ sao cho không tạo bọt quá mạnh khi đốt nóng (thường yêu cầu 5 đến 10 phút), tiếp theo đốt nóng cho đến khi bắt đầu than hóa. Từ từ thêm từng lượng nhỏ HNO3 đặc vào hỗn hợp, kiểm soát tốc độ phản ứng bằng điều khiển gia nhiệt trong quá trình thêm HNO3. Không đun quá mạnh để tránh quá trình than hóa xảy ra quá nhanh hoặc có thể gây mất As. Tiếp tục xử lý cho đến khi dung dịch có màu vàng nhạt xám. Nếu dung dịch vẫn tiếp tục có màu thì thêm 0,5ml dung dịch HClO4 và một ít HNO3 đặc đun nóng trong khoảng 15 phút, tiếp tục thêm 0,5ml dung dịch HClO4 và đốt nóng vài phút nữa. Ghi lại tổng lượng HNO3 sử dụng. Để nguội và pha loãng với 10ml nước. Dung dịch sẽ trong không màu (nếu có mặt Fe dung dịch có màu vàng nhạt). Đun sôi nhẹ cẩn thận tránh bọt cho đến khi có khói trắng xuất hiện. Tiếp tục để nguội và thêm 5ml nước đun sôi nhẹ tới khi có khói. Cuối cùng làm nguội và thêm 10ml HCl 5N đun sôi nhẹ vài phút. Để nguội và chuyển dung dịch vào bình định mức 50ml, tráng rửa bình Kjeldahl với lượng nhỏ của nước cất, gộp dịch rửa vào bình định mức. Định mức dung dịch tới mức 50ml bằng nước cất. Dung dịch này là dung dịch thử (Dung dịch A).
Chuẩn bị mẫu trắng:
Sử dụng các hóa chất và thuốc thử với lượng như đã dùng cho vô cơ hóa mẫu và xử lý như mẫu thử.
Xác định hàm lượng Sb, Ba, Cd, Cr, Cu, Pb, Zn bằng kỹ thuật AAS - ngọn lửa
Điều kiện của thiết bị
Chọn bước sóng đo và nhiên liệu đốt theo chỉ dẫn sau:
| Nguyên tố phân tích | Bước sóng - Vạch phổ (nm) | Nhiên liệu đốt tạo ngọn lửa |
| Sb | 217,6 | Không khí/C2H2 |
| Cd | 228,8 | Không khí/C2H2 |
| Cr | 357,9 | N2O/C2H2 |
| Cu | 324,8 | Không khí/C2H2 |
| Pb | 283,3 | Không khí/C2H2 |
| Zn | 213,9 | Không khí/C2H2 |
- Khe đo 0,2 - 0,5nm (tuỳ theo từng máy AAS, nhà sản xuất sẽ có chỉ dẫn cụ thể);
- Cường độ dòng đèn 70% giá trị của đèn HCL;
- Tốc độ khí (tuỳ theo từng máy AAS, nhà sản xuất sẽ có chỉ dẫn tốc độ khí tối ưu);
- Các điều kiện khác đạt theo chỉ dẫn đối với từng máy AAS cụ thể.
Tiến hành đo:
Đặt điều kiện đo cho máy AAS theo các thông số đã chọn. Tiến hành đo đối với chuẩn có nồng độ cao nhất để xác định thang cho đồ thị chuẩn. Tiếp tục đo với các chuẩn còn lại. Vẽ đồ thị chuẩn biểu thị mối tương quan giữa độ hấp thụ quang với nồng độ dung dịch chuẩn.
Tiến hành đo dung dịch thử và mẫu trắng.
Sử dụng đồ thị chuẩn để tính nồng độ kim loại cần đo trong dung dịch thử.
Tính kết quả:
| Nồng độ kim loại trong dung dịch thử (μg/ml) x 50 | = | Hàm lượng kim loại trong mẫu (mg/kg) |
| Khối lượng mẫu thử (g) |
Xác định hàm lượng As bằng kỹ thuật AAS - hydrid hóa
As được hydrid hóa trong thiết bị tạo khí hydrid chuyên dụng, bộ phận này được kết nối với máy AAS. Khí hydrid kim loại được dòng khí Ar đưa vào ngọn lửa hydro để nguyên tử hóa và đo phổ AAS.
Thiết bị đo:
Máy quang phổ hấp thụ nguyên tử với đèn catot rỗng phù hợp với bước sóng As 193,7nm
Dung dịch chuẩn Asen:
Dung dịch chuẩn gốc As 1000mg/l.
Pha dung dịch chuẩn trung gian: 5mg/l.
Pha dung dịch chuẩn làm việc: Dùng buret thêm lần lượt 0; 1; 2; 3; 4 và 5ml dung dịch chuẩn trung gian As vào các bình định mức 100ml và pha loãng với 50ml nước cất. Thêm 8ml H2SO4 đặc và 10ml HCl, lắc đều, định mức tới vạch với nước cất.
Dung dịch này có nồng độ As: 0; 0,05; 0,1; 0,15; 0,2; và 0,25μg/ml.
Xây dựng đường chuẩn:
Lấy 5ml dung dịch chuẩn làm việc nồng độ 0,25μg/ml vào trong bình hydrid hóa. Thêm 25ml nước cất và 2ml HCl 5N. Đậy nút lọ và đuổi hết không khí (kỹ thuật cụ thể theo chỉ dẫn của nhà sản xuất thiết bị), tạo môi trường khí Ar trong bình phản ứng. Ghép nối bộ hydrid hóa với bộ nguyên tử hóa của máy AAS bằng hệ thống ống dẫn khí có van by-pass. Khóa van by pass và cho NaBH4 vào bình phản ứng. (kỹ thuật cho NaBH4 vào bình phản ứng theo chỉ dẫn cụ thể đối với từng máy AAS do nhà sản xuất máy cung cấp). Đảm bảo hệ thống kín hoàn toàn. Khi phản ứng diễn ra chậm lại (20 - 30 giây), mở van by pass và van khí Ar đẩy hơi hydrid lên ngọn lửa nguyên tử hóa. Khi hơi hydrid đã qua hết (biểu thị bằng tín hiệu ghi), khóa van khí Ar, rửa bình hydrid hóa.
Đặt chế độ của máy để xác định thang đo phù hợp theo chuẩn nồng độ 0,25μg/ml. Tiếp tục đo với các chuẩn còn lại. Vẽ đồ thị chuẩn bị mối tương quan giữa độ hấp thụ quang với nồng độ dung dịch chuẩn.
Tiến hành:
Đo dung dịch thử và mẫu trắng. Sử dụng đồ thị chuẩn để tính nồng độ As cần đo trong dung dịch thử.
Tính kết quả:
| Nồng độ As trong dung dịch thử (μg/ml) x 50 | = | Hàm lượng As trong mẫu (mg/kg) |
| Khối lượng mẫu thử (g) |
XÁC ĐỊNH HÀM LƯỢNG TỔNG CÁC KIM LOẠI NẶNG: P.VC.10
Nguyên tắc:
Các kim loại nặng Pb, Hg, Cd, Sb, As, Ag, Cu, và một vài kim loại khác phản ứng với H2S tạo hợp chất sulfid có màu. Dựa trên cường độ màu của dung dịch xác định hàm lượng tổng các kim loại nặng quy về Pb2+.
Sn và Zn cũng tạo hợp chất sulfid nhưng tại pH = 3 - 4 chúng không có màu.
Hóa chất và thuốc thử:
Các hóa chất và thuốc thử phải là tinh khiết dùng trong phân tích.
- Dung dịch Ammoniac: Pha loãng 400ml NH4OH đặc tới 1000ml với nước cất.
- Acid hydrocloric (HCl) đậm đặc.
- Dung dịch chuẩn gốc Pb2+: sử dụng dung dịch nồng độ 1000mg/l.
- Dung dịch chuẩn làm việc Pb2+ nồng độ 10mg/l: pha khi sử dụng, pha loãng 1ml dung dịch chuẩn gốc với nước cất định mức đến 100ml.
Tiến hành:
Phương pháp 1
Dung dịch A: lấy 2ml dung dịch chì chuẩn (20μg Pb2+) vào trong ống nghiệm nessler 50ml. Điều chỉnh để pH nằm trong khoảng giữa 3 và 4 bằng cách thêm dung dịch acid acetic loãng hoặc dung dịch NH4OH, sau đó pha loãng với nước cất đến 40ml và khuấy đều hỗn hợp.
Dung dịch B: cho 25ml dung dịch thử (chuẩn bị theo chỉ dẫn trong các chuyên luận riêng) vào ống nghiệm nessler tương tự như ống dùng cho dung dịch A. Chỉnh pH dung dịch về 3,0 - 4,0 bằng dung dịch CH3COOH loãng TT.002 hoặc dung dịch NH4OH TT.009 (dùng giấy chỉ thị pH chính xác đo khoảng pH hẹp). Pha loãng tới 40ml bằng nước cất.
Dung dịch C: cho 25ml dung dịch thử (chuẩn bị theo chỉ dẫn trong các chuyên luận riêng) và thể tích (ml) dung dịch Pb2+ chuẩn tương đương với thể tích đã dùng để chuẩn bị dung dịch A vào ống nghiệm nessler tương tự như ống dùng cho dung dịch A và B. Chỉnh pH dung dịch về 3,0 - 4,0 bằng dung dịch CH3COOH loãng TT.002 hoặc dung dịch NH4OH TT.009 (dùng giấy chỉ thị pH chính xác đo khoảng pH hẹp). Pha loãng với 40ml bằng nước cất.
So sánh: (Tiến hành trong tủ hút) Thêm vào mỗi ống (dung dịch A, dung dịch B, dung dịch C) 10ml dung dịch H2S TT.131, trộn hỗn hợp kỹ rồi để yên 5 phút và đặt các ống xuống bề mặt màu trắng.
Đánh giá kết quả:
Màu dung dịch B là không được đậm hơn màu dung dịch A và màu dung dịch C tương đương hoặc đậm hơn màu dung dịch A.
Nếu màu của dung dịch C nhạt hơn màu của dung dịch A, phản ứng đã bị nhiễu, cần tiến hành phân tích lại theo phương pháp 2.
Phương pháp 2
- Chuẩn bị dung dịch A như trong phương pháp 1.
- Dung dịch B: Cân chính xác khối lượng mẫu (Theo chỉ dẫn trong các chuyên luận riêng) cho vào trong chén nung. Đưa chén vào tủ hốt, thêm H2SO4 đặc vào để làm ướt đẫm. Cẩn thận đốt cháy mẫu ở nhiệt độ thấp cho đến khi thành than, đậy chén nung bằng nắp (không đậy kín hoàn toàn) trong suốt quá trình đốt. Sau khi các chất đã hóa than hoàn toàn, thêm 2ml HNO3 và 5 giọt H2SO4 đặc, cẩn thận đốt nóng tiếp cho đến khi khói trắng bay ra. Tiếp theo đem nung trong lò nung ở 5000C đến 6000C cho đến khi tất cả cacbon cháy hết. Để nguội chén và thêm 4ml HCl loãng (1 trong 2), đậy nắp và phân hủy mẫu trong bể cách thủy 10 đến 15 phút. Mở nắp và làm bay hơi chậm trong bể cách thuỷ cho đến khô. Tẩm ướt cặn bằng 1 giọt HCl đặc, thêm 10ml nước nóng để trong 2 phút. Thêm từng giọt dung dịch NH4OH TT.009, thử bằng giấy quỳ, ngừng cho NH4OH TT.009 ngay khi hỗn hợp bắt đầu kiềm. Pha loãng dung dịch bằng nước tới 25ml và điều chỉnh pH của dung dịch về 3,0 - 4,0 bằng cách thêm CH3COOH loãng TT.002. Lọc dung dịch nếu cần, rửa chén nung và phễu lọc với 10ml nước rồi chuyển dung dịch lọc và dịch rửa vào ống nghiệm nessler dung tích 50ml, pha loãng với nước cất tới 40ml lắc kỹ hỗn hợp.
- Dung dịch C: tiến hành hóa mẫu như chuẩn bị dung dịch B. Thêm vào chén nung chứa mẫu thể tích (ml) dung dịch Pb2+ chuẩn tương đương với thể tích đã dùng để chuẩn bị dung dịch A.
So sánh: (Tiến hành trong tủ hút) Thêm vào mỗi ống (dung dịch A, dung dịch B, dung dịch C) 10ml dung dịch H2S TT.131, trộn hỗn hợp kỹ rồi để yên 5 phút và đặt các ống xuống bề mặt màu trắng.
Đánh giá kết quả:
Màu dung dịch B là không được đậm hơn màu dung dịch A và màu dung dịch C tương đương hoặc đậm hơn màu dung dịch A.
XÁC ĐỊNH TRO SULFAT: P.VC.11
Phương pháp 1 (đối với mẫu rắn)
Tiến hành:
Cân chính xác m (g) mẫu thử (khối lượng cân theo chỉ dẫn cụ thể trong các chuyên luận riêng) cho vào chén nung chịu nhiệt dung tích 50-100ml bằng Pt hoặc vật liệu chịu nhiệt phù hợp.
Tẩm ướt bằng dung dịch H2SO4 loãng TT.246, trộn đều. Đốt nóng nhẹ chén trên bếp điện (hoặc bộ đốt Argand, đèn đốt hồng ngoại) đốt cho đến khi mẫu khô và thành than đen hoàn toàn. Tiếp tục đốt cháy cho đến khi các thành phần trong mẫu hóa hơi và cặn than trong mẫu đã được oxy hóa hoàn toàn. Tẩm ướt cặn với 0,5ml dung dịch H2SO4 đặc, nung mẫu tại điều kiện như trên cho đến khi phần còn lại là cặn khô và H2SO4 dư hóa hơi hoàn toàn. Nung cặn trong lò nung ở nhiệt độ 8000C ± 250C trong 15 phút hoặc lâu hơn nếu cần thiết.
Làm nguội chén nung trong bình hút ẩm rồi xác định khối lượng cặn tro.
Chú ý:
Có thể cho lượng nhỏ (NH4)2CO3 vào trước khi kết thúc quá trình nung để thúc đẩy sự bay hơi của H2SO4.
Tính kết quả:
Hàm lượng tro sulfat (%) = x 100
Trong đó:
mx: khối lượng cặn thu được
m: khối lượng mẫu thử
Phương pháp 2 (đối với mẫu lỏng)
Tiến hành:
Nếu không có chỉ dẫn gì khác, cân chính xác m(g) mẫu thử (khối lượng cân theo chỉ dẫn cụ thể trong các chuyên luận riêng) cho vào chén nung thích hợp.
Thêm 10ml H2SO4 loãng TT.246, trộn đều. Đun nóng nhẹ (không đun sôi) để làm bay hơi mẫu thử đến khi còn cặn khô, làm nguội cặn. Nung cặn trong lò nung ở nhiệt độ 8000C ± 250C trong 15 phút hoặc lâu hơn nếu cần.
Làm nguội chén nung trong bình hút ẩm rồi xác định khối lượng cặn tro.
HƯỚNG DẪN PHƯƠNG PHÁP XÁC ĐỊNH CÁC THÀNH PHẦN HỮU CƠ: P.HC.
XÁC ĐỊNH CÁC HỢP CHẤT HỮU CƠ CƠ CLO: P.HC.01
Phương pháp so sánh độ đục
Chuẩn bị dung dịch mẫu thử:
Cân 0,25g mẫu (chính xác đến mg), cho vào bình nón (Bình A), hoà tan trong 10ml nước cất. Acid hóa bằng HNO3 và lọc lấy kết tủa. Trộn kết tủa thu được với 0,5g CaCO3, làm khô hỗn hợp và nung. Sau khi nung lấy cặn tro hòa tan trong 20ml dung dịch HNO3 loãng TT.170, lọc. Lấy phần dung dịch cho vào bình nón khác, thêm 0,5ml dung dịch AgNO3 0,1N, lắc đều.
Chuẩn bị dung dịch chuẩn so sánh:
Lấy dung dịch chuẩn HCl 0,01N (thể tích cụ thể theo chỉ dẫn trong các chuyên luận riêng) vào bình nón khác tương tự (Bình B). Thêm 20ml HNO3, thêm 0,5ml dung dịch AgNO3, lắc đều.
Đánh giá kết quả:
So sánh độ đục của 2 dung dịch. Độ đục của dung dịch trong bình A không được vượt quá độ đục của dung dịch trong bình B.
XÁC ĐỊNH CÁC HỢP CHẤT HỮU CƠ DỄ CACBON HÓA: P.HC.02
Chuẩn bị các dung dịch TSC (Colori-metric test solution):
- Dung dịch CuSO4 (TSC): Hoà tan 65g CuSO4.5H2O vào 25ml dung dịch HCl đặc và định mức bằng nước cất đến 1000ml.
- Hiệu chỉnh: Hút 10ml dung dịch CuSO4 TSC vào bình nón 250ml thêm 40ml nước cất và 3g KI, khuấy đều. Chuẩn iod tự do bằng dung dịch natri thiosulfat 0,1N dùng chỉ thị là dung dịch hồ tinh bột TT.242. 1ml dung dịch natri thiosulfat 0,1N tương đương với 24,97mg CuSO4.5H2O chỉnh lại thể tích của dung dịch CuSO4 (TSC) bằng HCl loãng sao cho 1ml dung dịch CuSO4 (TSC) có chứa 62,4mg CuSO4.5H2O.
- Dung dịch CoCl2 (TSC): Hoà tan 65g CoCl2.6H2O trong 25ml HCl đặc và định mức bằng nước cất đến 1000ml.
Hiệu chỉnh: Hút chính xác 5ml dung dịch CoCl2 (TSC) vào bình Iodin 250ml. Thêm 5ml dung dịch hydro peroxid TT.130 và 15ml dung dịch NaOH 20%. Đun trong 10 phút, để nguội. Cho thêm 2g KI và 20ml dung dịch H2SO4 20%. Chuẩn Iod tự do bằng dung dịch natri thiosulfat 0,1N dùng chỉ thị là dung dịch hồ tinh bột TT.242. 1ml dung dịch natri thiosulfat 0,1N tương đương với 23,8mg CoCl2.6H2O chỉnh lại thể tích của dung dịch CoCl2 (TSC) bằng dung dịch HCl loãng sao cho 1ml dung dịch CoCl2 (TSC) có chứa 59,5mg CoCl2.6H2O.
- Dung dịch FeCl3 (TSC): Hoà tan 55g FeCl3.6H2O trong 25ml HCl đặc và định mức bằng nước cất đến 1000ml.
Hiệu chỉnh: Hút chính xác 10ml dung dịch FeCl3 (TSC) vào bình Iodin 250ml. Thêm 5ml dung dịch hydro peroxid TT.130 và 15ml dung dịch NaOH 20%. Cho thêm 3g KI và 15ml nước cất, để yên 15 phút, pha loãng với 100ml nước cất. Chuẩn Iod tự do bằng dung dịch natri thiosulfat 0,1N dùng chỉ thị là dung dịch hồ tinh bột TT.242. 1ml natri thiosulfat 0,1N tương đương với 27,08mg FeCl3.6H2O chỉnh lại thể tích của dung dịch FeCl3 (TSC) bằng dung dịch HCl loãng sao cho 1ml dung dịch FeCl3 (TSC) có chứa 45,0mg FeCl3.6H2O.
Chuẩn bị dung dịch đối chứng:
Các dung dịch đối chứng được pha từ các dung dịch TSC theo tỷ lệ trong bảng dưới đây. Lần lượt dùng pipet lấy đúng tỷ lệ các dung dịch TSC, cho vào cốc thủy tinh, khuấy trộn đều.
Các dung dịch đối chứng
| Dung dịch đối chứng | Tỷ lệ dd CoCl2 (TSC) | Tỷ lệ dd FeCl3 (TSC) | Tỷ lệ CuSO4 (TSC) | Tỷ lệ nước cất | Màu |
| A | 0,1 | 0,4 | 0,1 | 4,4 | Vàng nâu nhạt |
| B | 0,3 | 0,9 | 0,3 | 8,5 | |
| C | 0,1 | 0,6 | 0,1 | 4,2 | |
| D | 0,3 | 0,6 | 0,4 | 3,7 | |
| E | 0,4 | 1,2 | 0,3 | 3,1 | Vàng đến vàng đỏ |
| F | 0,3 | 1,2 | 0,0 | 3,5 | |
| G | 0,5 | 1,2 | 0,2 | 3,1 | |
| H | 0,2 | 1,5 | 0,0 | 3,3 | |
| I | 0,4 | 2,2 | 0,1 | 2,3 | |
| J | 0,4 | 3,5 | 0,1 | 1,0 | |
| K | 0,5 | 4,5 | 0,0 | 0,0 | |
| L | 0,8 | 3,8 | 0,1 | 0,3 | |
| M | 0,1 | 2,0 | 0,1 | 2,8 | Vàng xanh |
| N | 0,0 | 4,9 | 0,1 | 0,0 | |
| O | 0,1 | 4,8 | 0,1 | 0,0 | |
| P | 0,2 | 0,4 | 0,1 | 4,3 | Hồng nhạt |
| Q | 0,2 | 0,3 | 0,1 | 4,4 | |
| R | 0,3 | 0,4 | 0,2 | 4,1 | |
| S | 0,2 | 0,1 | 0,0 | 4,7 | |
| T | 0,5 | 0,5 | 0,4 | 3,6 |
Chuẩn bị dung dịch mẫu:
Nếu không có chỉ dẫn gì khác, cân mẫu (khối lượng mẫu thử theo chỉ dẫn trong các chuyên luận riêng), nghiền mẫu thành dạng bột. Cho bột mẫu vào cốc thủy tinh không màu, chịu acid, có chứa sẵn 1 lượng dung dịch H2SO4 TT.245 (thể tích cụ thể của dung dịch H2SO4 theo chỉ dẫn trong các chuyên riêng). Khuấy đều hỗn hợp bằng đũa thủy tinh cho đến khi mẫu tan hoàn toàn. Để yên dung dịch trong 15 phút.
Nếu mẫu khó tan, cần đun nóng để hòa tan mẫu trong dung dịch H2SO4 TT.245, trộn mẫu và acid trong ống nghiệm, đun nóng theo chỉ dẫn trong các chuyên luận riêng. Khi mẫu đã tan hết, để nguội, chuyển dung dịch mẫu thử sang ống so sánh.
Nếu không có hướng dẫn gì khác, so sánh màu của dung dịch thử với màu của dung dịch đối chứng (dung dịch đối chứng cụ thể theo chỉ dẫn trong các chuyên luận riêng). Khi so sánh 2 dung dịch này phải được đựng trong ống so sánh bằng thủy tinh trong, không màu, cùng kích cỡ và đặt trên nền trắng.
XÁC ĐỊNH CÁC HỢP CHẤT KHỬ (TÍNH THEO GLUCO): P.HC.03
Tiến hành:
Cân 7g mẫu thử vào cốc có mỏ dung tích 400ml, hòa tan trong 35ml nước cất. Thêm 25ml dung dịch CuSO4 TT.079 và 25ml dung dịch tartrat kiềm TT.250. Đậy cốc bằng nắp hộp lồng. Đun nóng hỗn hợp sao cho thời gian đun đến sôi khoảng 4 phút và thời gian sôi là 2 phút. Lọc kết tủa Cu2O qua phễu lọc Gooch (phễu lọc đã được rửa sạch bằng nước nóng, tráng etanol, ete và sấy tại 1000C trong 30 phút). Rửa kết tủa thu được bằng nước cất nóng, 10ml etanol, cuối cùng rửa bằng 10ml ete. Sấy kết tủa tại 1000C trong 30 phút. Cân lượng Cu2O thu được.
Đánh giá kết quả:
Theo quy định cụ thể trong các chuyên luận riêng.
HƯỚNG DẪN PHƯƠNG PHÁP XÁC ĐỊNH ĐỐI VỚI PHẨM MÀU THỰC PHẨM: P.PM.
ĐỊNH TÍNH CÁC CHẤT MÀU: P.PM.01
Nhiều chất màu thực phẩm được sử dụng trong công nghiệp chế biến thực phẩm ở dạng hỗn hợp các chất màu thực phẩm như mô tả trong các chuyên luận, đôi khi trong hỗn hợp có thêm chất độn. Tiến hành phép thử đơn giản để xác định mẫu phẩm màu dạng bột là một chất màu đơn hay là một hỗn hợp của nhiều phẩm màu. Rải một lượng bột màu nhỏ vào 2 cốc, một cốc chứa nước, một cốc khác chứa acid sulfuric đặc. Dưới điều kiện này những hạt nhỏ của mỗi thành phần màu trong mẫu có thể quan sát được dễ dàng khi chúng hoà tan, phép thử này khá nhạy.
Việc xác định sự có mặt các chất màu thực phẩm riêng biệt thường khó khăn. Khá nhiều chất màu là các muối natri của acid sulfonic nên chúng không có điểm chảy và điểm sôi chính xác. Bên cạnh đó các chất màu tổng hợp thường có chứa các chất màu phụ trong khi các chất màu có nguồn gốc từ nguồn gốc thiên nhiên nhìn chung có chứa một số màu cơ bản khác nhau. Do đó nhận biết tốt nhất là bằng cách so sánh các tính chất quan sát được với các đặc tính của mẫu thương phẩm thật.
Các kỹ thuật chính được sử dụng là sắc ký và quang phổ, nhưng thường kết hợp cả 2 phương pháp. Ví dụ có mặt các chất màu phụ có thể ảnh hưởng đến đọc phổ do đó khó xác định định tính các chất màu chính, nên tách các chất màu bằng sắc ký cột, sắc ký giấy hay sắc ký lớp mỏng trước khi dùng các phương pháp nhận biết khác.
Sắc ký giấy và sắc ký lớp mỏng thường rất hữu ích trong việc nhận biết các chất màu, mà không cần đến các thiết bị đắt tiền. Nhưng luôn phải chú ý giá trị Rf của một chất chỉ là hàng số lý thuyết. Trong thực nghiệm có nhiều yếu tố ảnh hưởng, làm giá trị Rf trở nên không ổn định. Các yếu tố đó bao gồm: thành phần và thời hạn pha chế của hỗn hợp dung môi pha động, nồng độ của hơi dung môi trong không khí, chất lượng giấy, máy móc dụng cụ, loại và chất lượng của các chất phụ, nồng độ, giá trị pH của dung dịch và nhiệt độ. Vì lý do đó, sắc ký so sánh luôn luôn cần sử dụng. Bằng cách cho chạy đồng thời một vài chất với nồng độ như nhau, một số yếu tố ảnh hưởng này sẽ bị loại bỏ.
Sự trùng hợp về khoảng di chuyển của chất màu khi tiến hành sắc ký với một hệ dung môi đơn nhất định chỉ được coi là một chỉ tiêu nhận biết và các phép thử khác sẽ được tiến hành để khẳng định chắc chắn. Bảng sau đây cho những thí dụ về giá trị Rf của dung dịch 1% các chất màu trong nước, khai triển trên sắc ký mỏng silicagel G với 10 hệ dung môi khác nhau. Thành phần của các hệ dung môi này là (tất cả phải được chuẩn bị mới):
1. Iso-propanol: dung dịch amoniac (tỷ trọng 0,880): nước (7:2:1)
2. Iso-butanol: etanol: nước: dung dịch amoniac (tỷ trọng 0,880) (10:20:10:1)
3. Dung dịch kali nitrat bão hoà trong nước.
4. Phenol: nước (4:1-m/v)
5. Acid hydrocloric (tỷ trọng 1,180): nước (2g:15ml:85ml)
6. Trinatri citrat: dung dịch amoniac (tỷ trọng 0,880): nước (2g: 15ml: 85ml)
7. Aceton: etymetyl ketone: dung dịch amoniac (tỷ trọng 0,880): nước (60:140:1:60)
8. n-butanol: etanol: pyridin: nước (2:1:1:2)
9. iso-propanol: dung dịch amoniac (tỷ trọng 0,880) (4:1)
10. n-butanol: acid acetic (băng) : nước (10:5:6).
Những giá trị Rf của một số phẩm màu được phép và không được phép tan trong nước
| Tên phẩm màu | C1 N0 | EEC serial N0 | Dung môi số (xem mục 3-1) | |||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |||
| Màu đỏ | 16255 | E124 | 0,66 | 0,75 | 0,88 | 0,03 | 0,95 | 1,00 | 0,60 | 0,90 | 0,11 | 0,52 |
| Ponceau 4R Hay cochineal Rad A |
|
|
(0,85) |
|
|
|
|
|
|
|
|
(0,00-0,57) |
| Carmoisine Hay Azolubine | 14720 | E122 | 0,65 (0,77) | 0,81 | 0,00-0,42 | 0,16 | 0,00 (0,00-0,32) | 1,00 | 0,65 | 0,88 | 0,34 (0,46) | 0,63 (0,11-0,70) |
| Amaranth | 16185 | E123 | 0,62 (0,48-0,76) | 0,75 (0,83) | 1,00 (0,00-1,00) | 0,04 (0,16) | 1,00 | 1,00 | 0,40 (0,64-0,66) | 0,90 | 0,10 (0,41) | 0,39-0,67 |
| Erythrosine Hs | 45430 | E127 | 0,85 (068-0,79) | 0,91 (0,86-0,74-0,81) | 0,00-0,10 | 0,00-0,90 (0,41) | 0,00 | 0,00-0,95 | 0,64-0,66 (0,58) | 0,89 | 0,66 (0,57-0,43) | 1,00 |
| Red 2G | 18050 |
| 0,68 | 0,80 | 0,37 | 0,12 | 0,00-0,71 | 1,00 | 0,64 | 0,90 | 0,36 | 0,68 |
| Màu vàng cam |
|
|
|
|
|
|
|
|
|
|
|
|
| Orange G | 16230 |
| 0,71 (0,67-0,88) | 0,80 (0,75) | 0,64 (1,00-0,35) | 0,23-0,15 0,04 | 0,73 | 1,00 | 0,64 (0,62-0,50-0,67) | 0,91 | 0,36 (0,32-0,17) | 0,69 (0,46-0,82) |
| Orange R N | 15970 |
| 0,83 (0,62) | 0,88 (0,78) | 0,00 (0,00-0,42) | 0,42 (0,13) | 0,13 (0,38) | 0,76 (1,00) | 0,68 (0,65) | 0,92 | 0,64 (0,29) | 0,82-0,71 |
| Sunset yellow FCF Hay Orange yellow S | 15085 | E110 | 0,75 (0,68) | 0,82 (0,74) | 1,00 (0,00-1,00) | 0,17-0,03 | 1,00 | 1,00 | 0,65 (0,48) | 0,90 | 0,34 (0,10-0,22) | 0,67 (0,46) |
| Màu vàng |
|
|
|
|
|
|
|
|
|
|
|
|
| Tatrazine | 19140 | E102 | 0,66 | 0,77 | 0,46-1,00 | 0,08 | 1,00 | 1,00 | 0,52 | 0,93 | 0,14 | 0,50 |
| Yellow 2 G | 18965 |
| 0,63 | 0,80 | 0,77 | 0,21 | 0,74 | 1,00 | 0,62 | 0,92 | 0,21 | 0,75 |
| Quinoline Yellow | 47005 | E104 | 0,83-0,88 | 0,88 (0,82) | 0,00-1,00 | 0,65 (0,21) | 0,26-1,00 0,00-0,38 | 0,95 (0,35) | 0,54 (0,68) | 0,88 | 0,00-0,31 0,64 | 0,11-0,75 (0,83) |
| Fasi yellow AB | 13015 | E105 | 0,77 | 0,81 | 1,00 | 0,14 | 0,97 | 1,00 | 0,56 | 0,93 | 0,36 | 0,66 |
| Màu xanh |
|
|
|
|
|
|
|
|
|
|
|
|
| Green S hay axa | 44090 | E142 | 0,44 | 0,61 | 0,49 | 0,52 | 0,29 | 1,00 | 0,46 | 0,75 | 0,07 | 0,55 |
| Brillard green BS hay Lissamine green |
|
| (0,52-0,68-0,74) | (0,67-0,75-0,81-0,84) | (0,24) | (0,05-0,36-1,00) | (0,43) |
40,94 | (0,56-0,71) | (0,89-0,92) |
|
|
| Indigo Caranine hay indgotin | 73015 | E132 | 0,56 (0,70) | 0,50-0,76 (0,78) | 0,00 (0,05-0,90) | 0,09-0,18 (0,52) | 0,92 |
| 0,66 (0,71-0,73) | 0,89-0,84 | 0,37 (0,00-0,34) | 0,00-0,63 |
| Indanthrene Blue hay Solanthrene Blue RS Hay a..ragen Blue |
69800 |
E130 |
0,00 |
0,00 | 1,00 0,00 |
0,00 |
0,00 |
0,00 |
0,00 |
0,00 |
0,00 |
0,00 |
| Beillart blue FCF | 42090 |
| 0,64 (0,73) | 0,78 | 0,05 | 0,45 (0,68) | 0,10 | 0,00-1,00 | 0,61 (0,68) | 0,88 | 0,30 0,49-0,00-0,23) | 0,53 (0,64) |
| Patent Blue V | 42051 | E131 | 0,34-0,60 | 0,68 | 0,05 | 0,55 | 0,15 | 0,95 | 0,69 (0,72) | 0,84 (0,92) | 0,00-0,10 | 0,59 |
| Violet 6B | 42640 |
| 0,73 (0,67-0,91) | 0,80 (0,72) | 0,00 (0,00-0,46) | 0,62 (0,51-1,00) | 0,00-0,37 | 0,00-1,00 | 0,67 | 0,89 (0,62) | 0,37-0,45 0,70-0,76 | 0,64 (0,71) |
| Methyl violet | 42535 |
| 0,91 (0,80) | 0,56-0,81 0,90 | 0,00 (0,00-0,34) | 0,79-1,00 | 0,00-0,80 | 0,00 (0,00-0,53) | 0,00-0,68 (0,11-0,28) | 0,11 (0,90) | 0,94-0,87 (0,00-0,83) | 0,75-0,79 (0,00-0,07) |
| Màu nâu, đen |
|
|
|
|
|
|
|
|
|
|
|
|
| Brown FK |
|
| 0,78-0,71 0,66 | 0,79-0,86 | 1,00 (0,00-1,00) | 0,69-0,27 0,15-0,00 | 0,00-0,77 | 1,00 | 0,59-0,64 (0,53-0,37) | 0,93 | 0,34 (0,26-0,53) | 0,00-0,73 |
| Chocolate Brown FB |
|
| 0,00-0,69 | 0,00-0,75 | 0,00-0,82 | 0,00 (0,00-0,23) | 0,00-1,00 | 0,00-1,00 | 0,36-0,51 0,62 | 0,87 | 0,00-0,38 | 0,00-0,75 |
| Chocolate Brown HT | 202085 |
| 0,00-0,63 | 0,74 | 0,00-1,00 | 0,00 (0,00-0,16) | 0,00-1,00 | 0,00-1,00 | 0,34-0,43 0,62 | 0,88 | 0,00-0,32 | 0,00-0,73 |
| Black PN hay Brillant - Black BN | 28440 | E151 | 0,66 (0,47) | 0,75 | 1,00 (0,00-1,00) | 0,00 | 1,00 | 1,00 | 0,38 (0,61) | 0,85 | 0,05 | 0,00-0,43 |
| Black 7984 | 27755 | E152 | 0,62 | 0,75 | 1,00 | 0,00 | 1,00 | 1,00 | 0,38 (0,61) | 0,85 | 0,09 | 0,00-0,45 |
C1. No: số chỉ số phẩm màu “ ”; con số trong ngoặc chỉ các vết phụ của cường độ thấp hơn
O.x - O.y: kẻ vạch giữa các vết
Theo Pearson. D (1973), J.ASSOC Public. Anal, 11, 137-138 tái bản theo chất sinh ra ung thư do môi trường. Phương pháp phân tích tuyển chọn, tập 4 - Một số chất Amin thơm và thuốc nhuộm.
A3O trong tài liệu môi trường chung và môi trường công nghiệp (ấn phẩm IARC số 40) chi nhánh quốc tế về nghiên cứu ung thư, Lyon, 1981.
Việc đánh giá các vết màu cần làm trong khi sắc đồ vẫn còn ẩm dung môi, và cả sau khi sấy khô. Các vết màu sẽ được xem xét dưới tia sáng trắng, tốt hơn trong tia tử ngoại. Dưới tia tử ngoại (UV) nhiều chất màu có đặc tính thay đổi màu, hơn nữa các vết tạp chất huỳnh quang không màu trong mẫu có thể phát quang. Nếu có thể, sử dụng nguồn bức xạ UV với các bước sóng khác nhau; một đèn phát xạ ở khoảng 250nm.
Nên tiến hành các phép thử đối với acid, kiềm các thuốc thử thích hợp, để đảm bảo kết quả đúng. Tất cả các phép thử có thể được chấm vết bằng pipet mao quản trên mỗi vết màu.
Các yêu cầu sau đây phải đạt được khi định tính các chất màu:
- Các khoảng di chuyển Rf tương đương trong một vài hệ dung môi;
- Các vết màu tương đương dưới ánh sáng thường và ánh sáng tử ngoại;
- Sự thay đổi màu tương đương với các thuốc thử;
Việc kiểm tra bằng phương pháp quang phổ là phương pháp tốt nhất để xác định các chất màu. Các vùng tử ngoại, hồng ngoại và khả kiến đều được áp dụng.
Vùng khả kiến của quang phổ thông thường là phép kiểm tra trước tiên trong việc định tính một chất màu chưa biết. Có nhiều chất màu cho độ hấp thụ đặc trưng trong vùng khả biến, trong khi các chất màu khác thì không. Phổ trong vùng tử ngoại cũng có thể được sử dụng và nên thu nhận kết quả này, nếu có thể.
Phổ hấp thụ hồng ngoại thường là cách tốt nhất để xác định các hợp chất khác nhau, nhưng có một số khó khăn trong quá trình sử dụng phương pháp này.
Trong việc áp dụng phép đo quang phổ khả biến và tử ngoại, cần phải đo trong nhiều loại dung môi, hay nếu trong một loại dung môi, thì phải dưới những điều kiện khác nhau.
Phổ của nước cần được đo trong môi trường trung tính (đệm bằng amoni acetat), acid (acid hydrocloric 0,1N) và kiềm (natri hydroxid 0,1N).
Phổ hấp thụ được vẽ thành đồ thị biểu thị độ hấp thụ ở mọi bước sóng. Việc kiểm tra kết quả phổ cần bao gồm nhiều vùng của bước sóng hấp thụ cực đại. Toàn bộ đường phổ cần được xem xét cẩn thận để xác định dạng riêng biệt đường phổ từ “các gờ” hay các điểm uốn có thể là các nét đặc trưng và hữu ích nhất của phổ hấp thụ. Những nét đặc trưng này thường có thể phân biệt được giữa hai hoặc nhiều các chất màu có độ hấp thụ cực đại ở cùng bước sóng. Nhiều chất màu có thể xác định bằng việc quan sát phạm vi mà độ hấp thụ cực đại và những nét đặc trưng khác của đường cong phổ hấp thụ bị thay đổi trong các điều kiện pH khác nhau hoặc bởi có thay đổi dung môi.
Phổ hồng ngoại có thể đo được bằng nhiều cách, những cách thông dụng là:
- Phổ của dung dịch chất thử trong các dung môi thích hợp.
- Phổ của chất huyền phù trong 1 chất lỏng thích hợp.
- Kỹ thuật tạo viên kali bromid (một lượng nhỏ chất màu, thường 1- 3mg được trộn đều, kỹ với kali bromid khô, tinh khiết, hỗn hợp này được ép thành những viên trong dụng cụ chuyên dụng dưới áp suất nén từ 700 đến 1400 kg/cm2).
Phổ hồng ngoại được đo và ghi theo cách thông thường. Đặc trưng phổ là pic hấp thụ tại các bước sóng nhất định và dạng của các đường phổ gần kề những pic đó.
Việc đọc, giải phổ hồng ngoại cần phải thận trọng để đảm bảo chắc chắn rằng các pic hấp thụ do các chất tạp nhiễm vô cơ không được coi là pic hấp thụ của chất màu.
Cấu trúc tinh thể hoặc trạng thái vật lý khác của mẫu thử có thể ảnh hưởng đến phổ thu được từ huyền phù hay viên kali bromid. Cần đảm bảo chắc chắn rằng các chất chưa biết sẽ được xử lý chính xác theo đúng cách xử lý chất chuẩn hay mẫu đã biết. Các chất màu tan trong nước, thường có thể được xử lý bằng cách hoà tan trong nước, thêm một ít acid acetic, cho bốc hơi (cô) đến gần khô, và sau đó sấy khô ở khoảng 1000C để loại hết nước. Tất cả các chất màu được kiểm tra phải khan hoặc sạch dung môi khác trước khi đo phổ hồng ngoại, nước và tất cả các dung môi hữu cơ đều hấp thụ ánh sáng hồng ngoại.
Một thí dụ về việc sử dụng phổ hồng ngoại, có hai chất màu, 1 chất cho phép sử dụng (Sunset yellow) và một chất cấm sử dụng (Orange GGN) có phổ hấp thụ trong vùng khả biến và vùng tử ngoại gần giống nhau, khó có thể phân biệt được. Tuy nhiên, phổ hồng ngoại của chúng trong vùng phổ mà nhóm acid sulfonic hấp thụ mạnh hoàn toàn khác nhau.
Trong một vài trường hợp, phương pháp sắc ký, phương pháp quang phổ, và kết hợp của hai phương pháp trên có thể không cho kết quả đúng. Trong những trường hợp như vậy, vấn đề thường được giải quyết bằng cách giảm bớt chất màu hay phân hủy các hợp chất và xác định sản phẩm tạo ra. Kỹ thuật này đặc biệt được áp dụng cho các chất màu nhóm azo. Các hợp chất amin tạo ra từ sự phân giải có thể xác định ngay bằng kỹ thuật sắc ký và đo quang phổ.
XÁC ĐỊNH GIẢM KHỐI LƯỢNG KHI LÀM KHÔ: P.PM.02
Phẩm màu có chứa các nhóm SO3Na hay COONa thường có tính hút ẩm. Các phẩm màu thường giữ nước (có nguồn gốc từ nơi sản xuất hoặc từ môi trường) dưới dạng hydrat. Khi các sản phẩm phẩm màu được sấy ở 1350C, sự giảm khối lượng có thể tương đương với tổng hàm lượng nước, tuy nhiên điều này không luôn đúng với mọi trường hợp. Ví dụ Erythrosin và Ponceau 4R đều ngậm 1 phân tử nước ở dạng kết tinh tại 1350C. Thông thường phải lưu ý đến tính chất này khi tính tổng hàm lượng các chất tạo màu chính có trong mẫu thử.
Tiến hành:
Cân 2,0 - 3,0g mẫu phẩm màu trong chén có nắp có thể chịu được nhiệt độ 1350C, chén đã được cân bì (có thể sử dụng chén thủy tinh chuyên dụng xác định độ ẩm). Sấy đến khối lượng không đổi tại nhiệt độ theo quy định cụ thể trong các chuyên luận riêng, biên độ dao động nhiệt độ cho phép là ±50C. Cân khối lượng bì và mẫu sau khi sấy. Tính % khối lượng giảm đi sau khi sấy so với khối lượng mẫu thử.
XÁC ĐỊNH HÀM LƯỢNG CLORID (TÍNH THEO NaCl): P.PM.03
Thiết bị:
Sử dụng thiết bị chuẩn độ điện thế với điện cực chỉ thị bạc, điện cực so sánh calomel và cầu kali sunfat bão hòa.
Tiến hành:
Cân 0,5 - 1g mẫu thử phẩm màu, hòa tan trong 100ml nước cất, acid hóa bằng 5ml dung dịch HNO3 1,5N. Nhúng điện cực bạc vào dung dịch mẫu thử và nối điện cực calomel với dung dịch bằng cầu K2SO4 bão hòa. Có thể loại bỏ cầu K2SO4 bằng cách dùng một điện cực thủy tinh làm điện cực so sánh, điều này làm đơn giản hoá thiết bị, điện cực thủy tinh có độ ổn định cao được sử dụng để làm điện cực so sánh trong loại chuẩn độ này.
Xác định hàm lượng clorid trong dung dịch bằng cách chuẩn độ lại với dung dịch AgNO3 0,1N. Tính kết quả theo NaCl, 1ml dung dịch AgNO3 0,1N = 0,00585g NaCl.
Tính kết quả:
Hàm lượng clorid (%) tính theo NaCl = x 100
Trong đó:
V: thể tích dung dịch AgNO3 0,1N dùng để chuẩn độ
m: khối lượng mẫu thử phẩm màu.
XÁC ĐỊNH HÀM LƯỢNG SULFAT (TÍNH THEO Na2SO4): P.PM.04
Tiến hành:
Cân 5g mẫu thử phẩm màu, chuyển vào trong bình tam giác 250ml và hòa tan trong khoảng 100ml nước, đun nóng trong nồi cách thuỷ khoảng 1 giờ. Thêm 35g NaCl (không chứa SO42-), đậy nút bình và khuấy liên tục trong 1 giờ. Làm nguội và chuyển dung dịch NaCl bão hòa vào trong bình định mức 250ml, và pha loãng tới vạch mức ở 200C. Lắc bình và lọc dung dịch qua giấy lọc khô. Lấy 100ml của dịch lọc cho vào trong cốc 500ml, pha loãng tới 300ml với nước và acid hóa bằng HCl, thêm 1ml HCl dư. Đun nóng dung dịch đến khi sôi và thêm lượng dư dung dịch BaCl2 0,25N (thêm từng giọt và khuấy). Trộn đều hỗn hợp và để yên trên bếp 4 giờ hoặc để qua đêm ở nhiệt độ phòng, tiếp tục nâng nhiệt độ lên khoảng 800C và để lắng kết tủa. Lọc kết tủa BaSO4, rửa kết tủa với nước nóng và nung tại nhiệt độ màu đỏ đậm tới khối lượng không đổi (chén nung đã được cân bì).
Xác định mẫu trắng để hiệu chỉnh khối lượng BaSO4 thu được.
Tính kết quả:
Hàm lượng sulfat (%) tính theo Na2SO4 = x 100
Trong đó:
mx: khối lượng BaSO4 đã hiệu chỉnh.
m: khối lượng mẫu thử phẩm màu.
XÁC ĐỊNH HÀM LƯỢNG CÁC CHẤT KHÔNG TAN TRONG NƯỚC: P.PM.05
Tiến hành:
Cân 4,5 - 5,5g mẫu thử phẩm màu (một số phẩm màu có độ tan nhỏ hơn 5g/200ml, trong trường hợp này khối lượng mẫu thử cụ thể được chỉ dẫn trong chuyên luận riêng) cho vào cốc có mỏ 500ml, thêm khoảng 200ml nước nóng (800C - 900C) khuấy tới khi hoà tan và để dung dịch nguội tới nhiệt độ phòng. Lọc dung dịch qua phễu lọc thủy tinh xốp G4 đã biết trước khối lượng (hoặc bằng giấy lọc khô đã được cân bì) và rửa bằng nước lạnh cho đến khi nước rửa không còn màu. Sấy khô phễu lọc và cặn ở 1350C cho tới khối lượng không đổi. Ghi lại khối lượng cặn thu được.
Tính kết quả:
Hàm lượng chất không tan trong nước (%) = x 100
Trong đó:
mx: khối lượng cặn thu được.
m: khối lượng mẫu thử phẩm màu.
XÁC ĐỊNH HÀM LƯỢNG CÁC CHẤT MÀU PHỤ: P.PM.06
Định nghĩa:
Các chất màu phụ là các chất màu được tạo ra trong quá trình sản xuất để bổ sung vào chất tạo màu chính (chất được đặt tên cho sản phẩm). Bất kỳ chất màu nào khác có mặt trong sản phẩm ngoài chất tạo màu chính và các chất màu phụ đều được coi là giả, thông thường có thể phát hiện được chúng trên sắc ký đồ khi kiểm tra các chất màu phụ. Việc đọc sắc ký đồ để phát hiện các chất màu giả thường đòi hỏi ít nhiều kinh nghiệm.
Xác định bằng sắc ký giấy
Nguyên tắc:
Các chất màu phụ được tách ra khỏi chất màu chính bằng cách sử dụng sắc ký giấy và được giải hấp riêng rẽ từ giấy sắc ký. Độ hấp thụ quang của mỗi phần chiết được đo tại bước sóng cực đại trong vùng khả biến.
Vì việc nhận biết từng chất màu phụ trong sản phẩm là không khả thi, nên có thể chấp nhận một phương pháp gần đúng cho kết quả % tổng các chất màu phụ trên lượng mẫu thử. Coi khả năng hấp thụ quang của từng chất màu phụ giống với độ hấp thụ quang của tổng các chất màu. Độ hấp thụ quang của các phần chiết được gộp chung, liên hệ với độ hấp thụ quang của mẫu thử và hàm lượng tổng các chất màu để tính hàm lượng các chất màu phụ. Phép tính này được coi là phép tính gần đúng (chấp nhận được) cho việc xác định các chất màu phụ.
Thiết bị: Hình 4, hình 5.
Hình 4: Chi tiết bình khai triển sắc ký
Hình 5: Sơ đồ bình khai triển sắc ký
- Bình khai triển sắc ký và các dụng cụ phụ trợ (hình 4, hình 5).
- Khung đỡ các bản giấy sắc ký.
- Khay dung môi khai triển sắc ký;
- Bản giấy sắc ký cỡ 20cm x 20cm
- Microsyring có thể bơm được 0,1ml với dung sai là ±0,002ml.
- Máy UV-VIS (hoặc VIS)
Tiến hành:
Trước khi xác định khoảng gần 2 giờ, đổ dung môi khai triển vào đáy của bình sắc ký 1 lớp dày khoảng 1cm. Đặt khay dung môi D vào vị trí và đậy kín nắp bình.
Chấm 0,1ml dung dịch mẫu 1% lên bản giấy sắc ký (chấm làm nhiều lần, tổng thể tích dung dịch chấm lên 1 vết là 0,1ml, sấy khô vết chấm rồi chấm tiếp, kỹ thuật chấm phải đảm bảo vết chấm càng nhỏ càng tốt) trong khoảng hình chữ nhật có kích cỡ 18cm x 7mm, giữ cho đầu microsyring tiếp xúc đều đặn với mặt giấy sắc ký.
Hình 6: Hướng dẫn vị trí chấm trên giấy sắc ký
Để bản giấy khô ở nhiệt độ phòng trong 1-2giờ (hoặc làm khô ở 500C trong 5 phút rồi để ở nhiệt độ phòng 15 phút). Đặt bản giấy sắc ký có chấm mẫu cùng với một bản giấy sắc ký không chấm mẫu (mẫu trắng) lên khung (C) đỡ bản giấy sắc ký.
Rót dung môi sắc ký vào khay (D) sao cho bề mặt dung môi thấp hơn vạch chấm mẫu trên bản giấy sắc ký khoảng 1cm. Lượng dung môi sử dụng tuỳ thuộc vào kích thước của bình khai triển và phải được xác định trước. Đặt khung (C) đỡ bản giấy sắc ký vào vị trí và đậy nắp. Để dung môi sắc ký khai triển lên phía trên vạch chấm mẫu khoảng 17cm, nhấc khung (C) đỡ bản giấy sắc ký ra đưa vào bình hút ẩm ở 50-600C trong 10-15 phút. Nhấc bản giấy sắc ký ra khỏi khung (C). Có thể tiến hành khai triển đồng thời một vài bản giấy sắc ký một lần (nếu cần).
Cắt từ giấy sắc ký những dải băng màu phụ và cắt dải băng có vị trí tương tự trên bản không chấm mẫu, cho các dải này vào các ống nghiệm riêng biệt. Thêm 5ml hỗn hợp nước và aceton (tỉ lệ 1/1 về thể tích) vào mỗi ống thử, khuấy trong 2-3 phút, thêm 15ml dung dịch NaHCO3 0,05N và lắc để trộn kỹ hỗn hợp. Lọc phần dịch chiết màu và mẫu trắng qua giấy lọc đường kính 9cm (giấy lọc thưa), xác định độ hấp thụ quang của phần dịch chiết màu tại bước sóng hấp thụ cực đại, sử dụng cuvet có nắp kín. Trừ độ hấp thụ quang của dung môi nền, độ hấp thụ quang của dung môi được xác định bằng độ hấp thụ quang của hỗn hợp dung môi gồm 5ml của dung dịch nước/aceton (tỉ lệ 1/1 theo thể tích) và 15ml dung dịch NaHCO3 0,05N.
Thực hiện lọc phần dịch chiết từ mẫu trắng và đo theo các bước tương tự như trên.
Dung dịch chuẩn:
Chuẩn bị dung dịch mẫu thử 1%, pha dung dịch có nồng độ L/100% (L: giới hạn các chất màu phụ, quy định trong các chuyên luận riêng). Chấm 0,1ml dung dịch này (theo kỹ thuật đã mô tả ở trên). Tiến hành khai triển sắc ký, sau đó làm khô trong bình hút ẩm ở 50-600C trong 10-15 phút. Cắt dải giấy sắc ký theo vết sắc ký, cắt dải tương tự trong giấy sắc ký mẫu trắng cho chuẩn. Xác định độ hấp thụ thực của dung dịch chuẩn As.
Tính kết quả:
% hàm lượng các chất màu phụ = x L x
Trong đó:
Aa, Ab, Ac, … An: tổng độ hấp thụ của các chất màu phụ đã trừ độ hấp thụ mẫu trắng.
D: tổng hàm lượng chất màu có trong mẫu thử.
L: giới hạn của các chất màu phụ.
Các hệ dung môi khai triển sắc ký:
1. Nước: dd amoniac (d=0,88): trinatri citrat (95ml:5ml:2g)
2. n-butanol:nước:etanol:dd amoniac (d=0,88) (600:264:135:6)
3. 2-butanol:aceton:nước (7:3:3)
4. 2-butanol:aceton:nước:dd amoniac (d=0,88) (700:300:300:2)
5. 2-butanol:aceton:nước:dd amoniac (d=0,88) (700:160:300:2)
6. n-butanol:acid acetic băng:nước (4:1:5). Lắc đều trong 2 phút, để yên cho tách lớp, lấy lớp trên làm dung môi khai triển sắc ký.
XÁC ĐỊNH HÀM LƯỢNG CÁC CHẤT HỮU CƠ NGOÀI CHẤT MÀU: P.PM.07
Phương pháp sắc ký lỏng phân giải cao (HPLC)
Các tạp chất hữu cơ ngoài các chất màu được tách bằng HPLC với kỹ thuật gradient nồng độ pha động. Định lượng bằng so sánh diện tích pic của chúng với diện tích pic thu được từ các dung dịch chuẩn đã biết. Các điều kiện mô tả trong phương pháp này là những chỉ dẫn chung, để đạt được hiệu quả tách tốt có thể có những thay đổi nhỏ nếu cần.
Điều kiện tiến hành sắc ký:
Thiết bị: Sắc ký lỏng phân giải cao có bộ phận gradient nồng độ pha động.
Detector: UV do được độ hấp thụ quang ở bước sóng 254nm
Cột: thép không gỉ dài 1m x đường kính trong 2,1mm
Chất nhồi: chất trao đổi ion bazo mạnh ở (SAX). Ví dụ: màng polymer methacrylat có tẩm 1% (theo khối lượng) NH4OH.
Nồng độ mẫu: 2% trong natri tetraborat 0,01M.
Thể tích mẫu bơm: 10μl
Hệ dung môi pha động
+ Dung môi thứ nhất: dung dịch natri tetraborat 0,01M
+ Dung môi thứ hai: natri tetraborat 0,01M/natri perclorat 0,1M.
Chế độ gradient nồng độ: Quy định riêng trong từng chuyên luận.
Tốc độ dòng: 1,0ml/phút. Nhiệt độ: nhiệt độ phòng.
Lựa chọn điều kiện thực nghiệm như chiều dài cột, loại chất nhồi cột, hệ dung môi và sử dụng các bước tạo cặp ion, có thể dẫn sự biến đổi trong đặc tính rửa giải chất, như thứ tự rửa giải và độ phân giải chất.
Phương pháp sắc ký cột
Thiết bị:
Máy quang phổ UV hoặc UV-VIS
Cột sắc ký bằng thủy tinh (hình 7)
Chuẩn bị cột sắc ký:
Trộn bột cellulo (Whatman) vào dung dịch (NH4)2SO4 25% (hàm lượng Fe thấp). Nếu sử dụng loại cellulo khác phải chọn loại có hàm lượng Fe rất thấp.
Đơn vị cm
Hình 7: Cột sắc ký thủy tinh
Kiểm tra cột:
Chuẩn bị cột như chỉ dẫn. Cho 200ml dung dịch (NH4)2SO4 25% chảy qua cột, độ hấp thụ quang của dung dịch này trong vùng bước sóng tử ngoại phải đủ nhỏ để tránh gây nhiễu quá trình phân tích. Dùng khoảng 75g cellulo cho vào 500ml. Đặt một tấm lưới thép không rỉ vào trong cột, tại chỗ thắt phía dưới cột. Rót lượng vừa đủ bột cellulo đã nhào vào ống để tạo lớp pha tĩnh trong cột đạt chiều cao cách miệng cột 5cm. Trong lúc nhồi cột, vỗ nhẹ thành cột để đảm bảo cột được nhồi tốt. Rửa cột bằng 200ml dung dịch rửa giải.
Tiến hành:
Cho 0,2g mẫu phẩm màu vào trong cốc có mỏ có thể tích thích hợp, hoà tan trong 20ml nước cất. Thêm khoảng 5g bột cellulo. Thêm 50g (NH4)2SO4 để muối hóa mẫu. Nạp hỗn hợp này vào cột sắc ký, tráng cốc bằng dung dịch (NH4)2SO4 25% cho nước tráng cốc vào cột. Để nước trong cột rút cho đến khi ngừng chảy hoặc gần như ngừng chảy.
Cho dung dịch (NH4)2SO4 25% vào cột với tốc độ tương đương với tốc độ dung dịch chảy ra. Thu 1200ml dịch rửa giải, theo 12 phân đoạn riêng từng ống nghiệm, mỗi phân đoạn là 100ml. Giữ lại cột và các chất trong cột đến khi kiểm tra xong phân đoạn rửa giải cuối cùng.
Lắc đều từng phân đoạn rửa giải thu được. Đo phổ hấp thụ quang UV của từng phân đoạn tại vùng bước sóng 220nm - 400nm. Nếu phổ hấp thụ quang của phân đoạn rửa giải cuối cùng cho thấy các chất chưa rửa giải hết ra khỏi cột (vẫn xuất hiện cực đại hấp thụ), tiếp tục tiến hành rửa giải các chất trong cột cho đến khi kiểm tra độ hấp thụ quang của dịch rửa giải cho kết quả các chất đã được rửa giải hoàn toàn.
Thông thường chỉ gặp 1 hợp chất, việc định tính hoặc định lượng chất đó được tiến hành bằng cách so sánh phổ hấp thụ quang của hợp chất được rửa giải ra với phổ hấp thụ quang của dung dịch chất tinh khiết trong cùng loại dung môi.
Trong trường hợp có mặt từ hai chất trở lên với lượng đáng kể trong bất kỳ phân đoạn nào, các dữ liệu quang phổ sẽ chỉ ra điều này. Đối với trường hợp này, hàm lượng các chất được xác định bằng quy trình trong phân tích quang phổ hỗn hợp nhiều chất hấp thụ quang.
Một số mẫu thử có chứa lượng nhỏ các chất khác nhau, đặc biệt là các muối vô cơ, điều đó gây nên “hấp thụ nền”. Hiệu chỉnh “hấp thụ nền” được tiến hành như sau:
Xác định lượng chất hấp thụ nền tại phân đoạn rửa giải ngay trước và ngay sau phân đoạn xác định chất màu phụ. Tính trung bình tổng độ hấp thụ của hai phân đoạn này (ATB). Lấy độ hấp thụ quang của phân đoạn xác định chất màu phụ trừ đi giá trị trung bình ATB, hiệu số còn lại được coi là độ hấp thụ quang của chất màu phụ cần kiểm tra.
XÁC ĐỊNH HÀM LƯỢNG CÁC CHẤT CÓ THỂ CHIẾT BẰNG ETE: P.PM.08
Phương pháp chiết soxhlet
Thiết bị
Bộ chiết soxhlet, treo 1 sợi dây Cu lơ lửng trên bầu ngưng của ống soxhlet và một miếng Cu nhỏ (0,5g) vào bình chưng cất.
Hoá chất:
Dietyl ete hoặc diisopropyl ete.
Tinh chế ete như sau: Ngay trước khi sử dụng, cho ete đã được cất lại qua cột sắc ký chứa pha tĩnh là oxid nhôm (loại dùng cho sắc ký) để loại peroxid và các chất ức chế. Kiểm tra để chắc chắn đã loại hết peroxid như sau:
Chuẩn bị dung dịch sắt (II) thiocyanat (trong, không màu) bằng cách trộn thể tích tương ứng dung dịch FeSO4 0,1N và dung dịch NH4SCN 0,1N. Loại bỏ màu đỏ trong dung dịch (nếu có, màu đỏ được tạc ra do sự có mặt của Fe3+) bằng TiCl3. Thêm 10ml ete vào 50ml dung dịch này, lắc kỹ trong 2-3 phút. Dung dịch không được xuất hiện màu đỏ.
Tiến hành:
Cân 2g mẫu thử phẩm màu. Chuyển vào ống chiết soxhlet, tiến hành chiết với 150ml ete trong 5 giờ. Cô đặc dịch chiết ete thu được trên bể cách thủy tới khi còn 5ml. Chuyển dung dịch thu được vào chén sấy đã cân bì, làm bay hơi hết ete trên bể cách thủy sau đó sấy tại 1050C đến trọng lượng không đổi. Để nguội trong bình hút ẩm. Cân và tính hàm lượng chất có thể chiết bằng ete so với mẫu thử.
XÁC ĐỊNH HÀM LƯỢNG CÁC HỢP CHẤT AMIN THƠM BẬC NHẤT KHÔNG SULPHONAT HÓA: P.PM.09
Nguyên lý:
Các hợp chất amin thơm bậc nhất không sulphonat hóa được chiết tách ra khỏi dung dịch mẫu thử kiềm tính bằng toluen, sau đó được chiết lại trong acid, tiến hành diazo hoá và tạo phức. Định lượng bằng cách đo độ hấp thụ quang (trong vùng khả biến) trên máy quang phổ kế. Kết quả được tính theo anilin trừ khi đã biết chúng là chất amin khác.
Thiết bị:
Máy quang phổ kế hai chùm tia đo được toàn vùng ánh sáng khả kiến.
Hóa chất, thuốc thử:
Các hóa chất thuốc thử phải tinh khiết phân tích
Toluen
Dung dịch HCl nồng độ (gần đúng) 1N; 3N
Dung dịch KI nồng độ (gần đúng) 50%
Dung dịch Na2CO3 nồng độ (gần đúng) 2N
Dung dịch NaOH nồng độ (gần đúng) 1N
Dung dịch NaOH nồng độ (gần đúng) 0,1N
Dung dịch muối R (Dinatri 2-naphtol-3,6-disulfonat) nồng độ (gần đúng) 0,05N
Dung dịch NaNO2 nồng độ (gần đúng) 0,5N
Nước cất
Chất chuẩn:
Dung dịch anilin chuẩn được pha như sau:
Dung dịch A: Cân chính xác 0,1g anilin đã cất lại vào cốc có mỏ loại nhỏ, cho vào bình định mức 100ml, tráng cốc vài lần bằng nước cất, thu nước tráng vào bình định mức chứa alinin, thêm 30ml dung dịch HCl (~3N), định mức đến vạch bằng nước cất. Giữ dung dịch tại nhiệt độ phòng.
Dung dịch B (chuẩn bị hàng ngày trước khi sử dụng): hút 10ml dung dịch A cho vào bình định mức 100ml, định mức đến 100ml bằng nước cất tại nhiệt độ phòng. 1ml dung dịch này tương đương với 0,0001g alinin.
Xây dựng đường chuẩn:
Lần lượt hút 5ml, 10ml, 15ml, 20ml, 25ml, cho vào các bình định mức 100ml khác nhau. Định mức đến 100ml bằng dung dịch HCl (~1N). Lần lượt hút 10ml mỗi dung dịch chuẩn vào các ống nghiệm sạch và khô, làm lạnh các ống nghiệm bằng cách nhúng vào nước đá trong 10 phút. Thêm vào mỗi ống 1ml dung dịch KI (~50%) và 0,5ml dung dịch NaNO3 (~0,5N), lắc đều và nhúng vào nước đá trong 10 phút. Lấy 5 bình định mức 25ml, cho vào mỗi bình 1ml dung dịch muối R (0,05N) và 10ml Na2CO3. Lần lượt rót từng ống nghiệm đang ngâm trong nước đá vào các bình định mức 25ml đã chuẩn bị sẵn. Rửa các ống nghiệm bằng vài giọt nước, thu nước rửa vào các bình định mức 25ml tương ứng. Định mức tới vạch mức 25ml bằng nước cất. Đậy nút bình lắc đều, sau đó để yên trong chỗ tối 15 phút.
Đo độ hấp thụ quang của từng dung dịch chuẩn tại bước sóng 510nm. Sử dụng mẫu trắng là hỗn hợp 10ml dung dịch HCl 1N; 10ml dung dịch Na2CO3; 2ml dung dịch muối R; định mức tới 25ml bằng nước cất. Vẽ đồ thị biểu thị mối tương quan giữa độ hấp thụ quang của dung dịch với nồng độ alinin trong dung dịch.
Tiến hành:
Cân khoảng 2g (chính xác đến 0,01g) mẫu thử phẩm màu cho vào phễu chiết chứa 100ml nước cất, lắc đều. Tráng thành phễu bằng 50ml nước cất nữa, lắc đều để mẫu tan hoàn toàn. Thêm 5ml dung dịch NaOH 1N. Chiết bằng 50ml toluen (2 lần). Gộp dịch chiết toluen và rửa dịch toluen bằng 10ml dung dịch NaOH 0,1N (nhiều lần) đến khi loại hết màu. Chiết dịch toluen với 10ml dung dịch HCl 3N (3 lần). Gộp dịch chiết HCl và định mức đến 100ml bằng nước cất, lắc đều, dung dịch này ký hiệu là dung dịch T.
Hút 10ml dung dịch T vào ống nghiệm sạch, khô. Làm lạnh 10 phút trong nước đá. Thêm 1ml dung dịch KI (~50%) và 0,5ml dung dịch NaNO3 (~0,5N), lắc đều và nhúng vào nước đá trong 10 phút. Lấy bình định mức 25ml, cho vào 1ml dung dịch muối R (0,05N) và 10ml Na2CO3, chuyển dung dịch trong ống nghiệm đang ngâm trong nước đá vào bình định mức 25ml đã chuẩn bị sẵn. Rửa ống nghiệm bằng vài giọt nước, thu nước rửa vào các bình định mức 25ml. Định mức tới 25ml bằng nước cất. Đậy nút bình lắc đều, sau đó để yên trong chỗ tối 15 phút.
Đo độ hấp thụ quang của dung dịch thử tại bước sóng 510nm. Sử dụng mẫu trắng là hỗn hợp 10ml dung dịch T; 10ml dung dịch Na2CO3; 2ml dung dịch muối R; định mức tới 25ml bằng nước cất. Từ độ hấp thụ quang của dung dịch đo được, đọc trên đồ thị chuẩn suy ra khối lượng alinin có trong dung dịch thử.
Hàm lượng các hợp chất amin thơm bậc nhất không sulphonat hóa được tính theo công thức sau:
Hàm lượng (%) =
XÁC ĐỊNH HÀM LƯỢNG LEUCO BASE TRONG CHẤT MÀU TRIARYL-METAN SULPHONAT HÓA: P.PM.10
Nguyên tắc:
Không khí cho qua dung dịch chất nước chứa clorid và dimetyl formamid, ở điều kiện này các leuco base bị oxi hóa tạo hợp chất màu. Sự gia tăng độ hấp thụ quang biểu thị tương quan với hàm lượng leuco base trong mẫu thử.
Thuốc thử:
Dimetylformamid (DMF)
Dung dịch A: Cân 10,0g CuCl2.2H2O và hoà tan trong 200ml DMF. Chuyển vào bình định mức 1000ml và định mức đến vạch bằng DMF.
Dung dịch B: Cân chính xác 110 ± 5g mẫu thử phẩm màu, hoà tan trong khoảng 100ml nước cất, chuyển vào bình định mức 1000ml và định mức tới vạch với nước cất.
Tiến hành:
Dung dịch I: Lấy 50ml DMF vào bình định mức 250ml, đậy nắp bằng giấy parafin và để ở chỗ tối.
Dung dịch II: lấy chính xác 10ml dung dịch B vào bình định mức 250ml. Thêm 50ml DMF. Đậy nắp bằng giấy parafin và để ở chỗ tối.
Dung dịch III: Lấy 50ml dung dịch A vào trong bình định mức 250ml, sục bọt khí vào dung dịch này trong 30 phút theo cách sau: Nối pipet 5ml vào trong hộp gắn với nguồn khí. Mở dòng khí từ từ. Cho đầu pipet vào dung dịch trong bình định mức và điều chỉnh dòng khí nhanh nhưng kiểm soát được. Sau 30 phút nhấc pipet ra khỏi dung dịch và cho nước rửa pipet vào bình định mức. Tắt dòng khí.
Dung dịch d1 và d2: Lấy chính xác 10ml dung dịch B vào trong hai bình định mức 250ml và thực hiện giống như dung dịch II. Thêm 50ml dung dịch A vào mỗi bình. Thổi không khí vào dung dịch trong 30 phút như phương pháp trên.
Sau 30 phút cho bọt khí qua dung dịch, định mức tất cả 5 bình gần đến vạch mức bằng nước cất. Khi trộn DMF vào nước dung dịch nóng lên, do vậy đặt các bình định mức trong bể điều nhiệt hoặc dưới vòi nước chảy cho đến khi chúng nguội đến nhiệt độ phòng, không để lâu hơn, khoảng 5 đến 10 phút là thích hợp. Định mức chính xác tới vạch mức bằng nước cất. Đưa ngay dung dịch vào máy đo phổ hấp thụ. Toàn bộ quá trình đo tiến hành càng nhanh càng tốt.
Tiến hành đo phổ: Quét phổ hấp thụ ở dải bước sóng từ 700 đến 500nm với cửa sổ quét 0,1nm (dùng cuvet đo quang 1cm). Đo phổ tất cả các dung dịch trên cùng đồ thị và lấy giá trị tại bước sóng cực đại giữa 620 và 635nm
Xác định mẫu thử:
Chú ý: Cuvet đo phải được rửa trước mỗi lần chạy. Đối với cuvet đo liên tục, rửa cuvet bằng 3 lần X 40ml dung dịch mẫu thử.
Mẫu so sánh
| Đường cong | Cuvet | Cuvet | Nhận xét |
| 1 | I | I | đặt điểm 0 tại 700nm, đo phổ, ghi kết quả tại độ hấp thụ chuẩn của chất màu |
| 2 | I | II | Thực hiện đo phổ không cần đặt về 0, ghi độ hấp thụ tại cực đại hấp thụ |
| 3 | III | III | đặt về 0 tại 700nm, đo phổ, ghi kết quả tại độ hấp thụ chuẩn của chất màu. |
| 4a | III | d1 | Thực hiện đo phổ không cần đặt về 0, ghi độ hấp thụ tại cực đại hấp thụ |
| 4b | III | d2 | Thực hiện đo phổ không cần đặt về 0, ghi độ hấp thụ tại cực đại hấp thụ |
d1 và d2 cần được đo 2 lần
Tính kết quả:
Hàm lượng leuco base (%) =
Trong đó:
a: độ hấp thụ của 100% chất màu
m: tổng khối lượng mẫu tính bằng mg.
t: tỷ lệ phân tử lượng (MW) chất màu/Phân tử lượng (MW) leuco base (hướng dẫn cụ thể trong các chuyên luận riêng).
A1, A2, A3, A4: độ hấp thụ lớn nhất của đường cong 1, 2, 3, 4 tương ứng
ĐỊNH LƯỢNG CÁC CHẤT MÀU CHÍNH: P.PM.11
Phương pháp xác định chất màu bằng quang phổ UV-VIS
Nguyên tắc:
Độ hấp thụ của dung dịch chất màu được đo tại bước sóng hấp thụ cực đại và so sánh với độ hấp thụ của dung dịch chuẩn.
Thiết bị:
Máy đo phổ hấp thụ UV-VIS có độ chính xác cao (dung sai ± 1% hoặc ít hơn) có thể đo trong vùng bước sóng 350 - 700nm với độ rộng khe sáng là 10nm hoặc nhỏ hơn.
Cuvet đo phổ hấp thụ có độ dày truyền sáng 1cm
Hóa chất:
Nước cất (mới cất)
Các hóa chất khác được chỉ dẫn cụ thể trong các chuyên luận riêng.
Phương pháp 1:
Tiến hành:
Cân chính xác 0,25g (± 0,02g) mẫu thử, chuyển vào bình định mức 1000ml. Thêm nước cất hoặc dung môi hòa tan theo chỉ dẫn cụ thể trong các chuyên luận riêng, lắc trộn hỗn hợp, định mức đến vạch. Pha loãng dung dịch này đến nồng độ theo chỉ dẫn cụ thể trong các chuyên luận riêng. Đo độ hấp thụ quang (A) của dung dịch tại bước sóng hấp thụ cực đại.
Tính kết quả:
% tổng chất màu = x 100
Trong đó:
A: độ hấp thụ của dung dịch mẫu thử
A1%1cm: độ hấp thụ riêng (chỉ dẫn trong các chuyên luận riêng)
Vol: hệ số pha loãng (thể tích pha loãng/thể tích đo)
W: trọng lượng mẫu thử
Phương pháp 2:
Tiến hành:
Cân khoảng 0,08g mẫu (w) cho vào định mức 100ml (V1) hoà tan trong 20ml CHCl3 (tinh khiết không có acid). Bảo đảm rằng dung dịch này phải trong, mẫu phải tan hoàn toàn. Định mức đến vạch bằng cyclohexan - Dung dịch F1. Hút 5ml dung dịch F1 vào bình định mức 100ml (V2), định mức đến vạch bằng cyclohexan - Dung dịch F2. Tương tự hút 5ml dung dịch F2 vào bình định mức 100ml, định mức đến vạch bằng cyclohexan - Dung dịch F3. Đo độ hấp thụ quang của dung dịch F3 sử dụng mẫu trắng là cyclohexan.
Tính kết quả:
Hàm lượng chất tạo màu chính (%) =
Trong đó:
A: độ hấp thụ của dung dịch F3 tại bước sóng hấp thụ cực đại
A1%1cm: độ hấp thụ riêng của chất chuẩn (chỉ dẫn trong các chuyên luận)
V1: thể tích bình định mức thứ nhất (100ml)
V2: thể tích bình định mức thứ hai (100ml)
V3: thể tích bình định mức thứ ba (100ml)
v1: thể tích dung dịch F1 lấy để pha loãng (5ml)
v2: thể tích dung dịch F2 lấy để pha loãng (5ml)
w: khối lượng mẫu thử (g).
Chú ý:
Tiến hành chuẩn bị các dung dịch đồng thời, đo quang ngay khi chuẩn bị xong các dung dịch. Tiến hành thí nghiệm trong điều kiện tránh ánh sáng và tránh tiếp xúc với không khí.
Phương pháp chuẩn độ với Titan clorid
Thiết bị:
Thiết bị sinh CO2, bình Kip
Chuẩn bị thiết bị chuẩn độ: (hình 8)
Chuẩn bị dung dịch Titan clorid 0,1N:
Cho vào một bình tam giác cỡ lớn lượng dung dịch titan clorid 15% sao cho trong 1 lít dung dịch cần chuẩn bị có chứa 15,5 - 16,5 titan clorid. Thêm HCl đặc (tỷ trọng 1,16 - 1,18) vào dung dịch theo tỷ lệ 100ml HCl cho 1 lít dung dịch cần chuẩn bị. Đun sôi hỗn hợp trong 1-2 phút, sau đó rót hỗn hợp này vào nước cất đã được làm lạnh. Thêm nước cất đến thể tích cần thiết trong aspirator, lắc kỹ, bảo quản dung dịch trong môi trường khí CO2. Bảo quản dung dịch trong lọ dưới một lớp dầu parafin (dày khoảng 0,6cm)
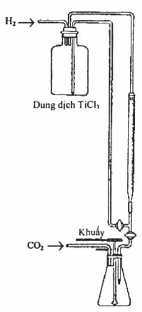
Hình 8: Sơ đồ thiết bị chuẩn độ
Chuẩn hóa dung dịch Titan clorid:
Cân 3g muối amoni fero sulfat [(NH4)SO4.FeSO4.6H2O] cho vào một bình nón 500ml, cho khí CO2 đi qua bình liên tục cho đến khi kết thúc quá trình xác định. Thêm 50ml nước cất và 25ml dung dịch H2SO4 10N, sau đó thêm 30ml dung dịch kali dicromat 0,1N, Chuẩn độ với dung dịch titan clorid đến khi gần tới điểm tương đương đã tính trước. Thêm 5ml dung dịch NH4SCN 20% và tiếp tục chuẩn độ đến khi màu đỏ biến mất và dung dịch bắt đầu chuyển sang màu xanh. Xác định một mẫu trắng với cùng điều kiện như trên. Hệ số bổ chính cho dung dịch titan clorid 0,1N được tính theo công thức:
F = 30/thể tích chính xác (ml) của dung dịch titan clorid sử dụng để chuẩn độ.
Tiến hành xác định tổng hàm lượng chất màu trong mẫu thử:
Cân chính xác mẫu thử phẩm màu (khối lượng chính xác theo chỉ dẫn trong chuyên luận riêng), cho vào bình thủy tinh dung tích 500ml, thêm 10g natri citrat hoặc 15g natri hydro tartrat và 150ml nước cất. Cho khí CO2 qua bình thủy tinh, đun sôi dung dịch. Chuẩn độ dung dịch với dung dịch titan clorid đã chuẩn hóa ở trên, giữ dòng khí CO2 ổn định. Gần đến điểm tương đương, thêm từng giọt dung dịch titan clorid đến điểm tương đương. Lúc này chất màu đóng vai trò là chất chỉ thị của chính bản thân nó, trừ khi có chỉ dẫn khác trong các chuyên luận riêng.
Tính kết quả:
Tổng hàm lượng chất màu (%) =
Trong đó:
V: thể tích dung dịch titan clorid 0,1N (dùng để chuẩn độ)
D: Khối lượng chất màu tương đương với 1ml dung dịch TiCl2 0,1N (chỉ dẫn cụ thể trong từng chuyên luận riêng).
CÁC DUNG DỊCH, THUỐC THỬ TT
Chú ý:
Các hóa chất sử dụng để pha các dung dịch dưới đây đều phải đạt chất lượng “Tinh khiết phân tích”.
PbT: dung dịch không có Pb.
Dung dịch acid acetic (dung dịch acid acetic đặc) TT.001
Dung dịch CH3COOH trong nước nồng độ ~30% - (m/v) (~5N).
Dung dịch acid acetic loãng TT.002
Dung dịch CH3COOH trong nước nồng độ ~6% - (m/v) (~1N).
Dung dịch anhydrid/benzol TT.003
Dung dịch gồm: 10ml anhydrid acetic và 90ml benzol
Dung dịch anhydrid acetic/pyridin TT.004
Thêm pyridin khan vào 25g anhydrid acetic, định mức đến 100ml bằng pyridin khan. Dung dịch sử dụng trong ngày.
Dung dịch acid acetic periodic TT.005
Hoà tan 5,4g acid periodic trong 100ml nước cất, thêm 1900ml acid acetic băng, trộn đều. Giữ dung dịch trong lọ kín thuỷ tinh màu hoặc lọ thủy tinh trong nhưng trong chỗ tối.
Dung dịch alizarin vàng GG TT.006
Hoà tan 0,1g alizarin vàng GG trong 100ml etanol. Lọc nếu cần
Dung dịch alizarin vàng GG/thymol-phtalein TT.007
Trộn 10ml dung dịch alizarin vàng GG TT.006 với 20ml dung dịch thymolphtalein TT.253.
Dung dịch acid 1-amino-2-naphtol-4-sulfonic TT.008
Hoà tan 0,2g acid 1-amino-2-naphtol-4-sulfonic trong 195ml dung dịch Natri bisulfit (3 trong 20) và 5ml natri sulfit (1 trong 5), lọc nếu cần. Đóng nắp, giữ lạnh, trong chỗ tối. Dung dịch dùng trong 10 ngày sau khi pha.
Dung dịch amoniac (Dung dịch amoniac loãng) TT.009
Dung dịch NH3 trong nước nồng độ 9,5% - 10,5% (m/v) (~6N). Pha loãng 400ml dung dịch NH4OH 28% tới 1000ml bằng nước cất.
Dung dịch amoniac đặc TT.010
Dung dịch NH3 trong nước nồng độ ~25% - m/v (~15N).
Dung dịch amoniac loãng (PbT) TT.011
Là dung dịch amoniac TT.009 đạt yêu cầu sau: Lấy 20ml dung dịch amoniac TT.009 thêm 1ml dung dịch Kali cyanid (PbT) TT.193, định mức đến 50ml bằng nước cất và thêm 2 giọt Natri sunfit (PbT) TT.239. Dung dịch không được thẫm màu lại.
Dung dịch amoniac trong etanol TT.012
Dung dịch NH3 trong etanol. Dung dịch trong, không màu có mùi amniac. Trọng lượng riêng khoảng 0,8. Giữ dung dịch trong lọ nút cao su, giữ lạnh.
Dung dịch đệm amoni/amoni clorid (pH~10) TT.013
Hòa tan 67,5g amoni clorid (NH4Cl) trong nước, thêm 570ml amoni hydroxid 28% định mức đến 1000ml bằng nước cất.
Dung dịch amoni bạc nitrat TT.014
Thêm (nhỏ giọt) dung dịch amoniac TT.009 vào dung dịch bạc nitrat (1 trong 20) đến khi vừa tạo kết tủa, nhưng kết tủa không bền, hoà tan ngay. Lọc dung dịch, bảo quản trong lọ thủy tinh màu.
Chú ý: Dung dịch amoni bạc nitrat sẽ tạo hợp chất gây nổ khi bảo quản yên, không giữ dung dịch này lâu, pha ngay khi dùng. Khi kết thúc công việc, rửa ngay dụng cụ, trung hòa dung dịch amoni bạc nitrat bằng HCl.
Dung dịch amoni acetat TT.015
Dung dịch amoni acetat 10% (CH3COONH4) trong nước.
Dung dịch amoni acetat citrat (PbT) TT.016
Hoà tan 12,5g amoni acetat (CH3COONH4) và 12,5g amoni citrat [C3H4OH(COOH)(COONH4)2] trong nước. Thêm dung dịch amoniac đặc TT.010 cho đến khi dung dịch có môi trường kiềm (thử bằng giấy thymol xanh) thêm nước đến 100ml. Tinh chế bằng dung dịch dithizon trong clorofom, lắc dung dịch với clorofom bảo quản loại hết dithizon dư.
Dung dịch amoni carbonat TT.017
Hoà tan 20g amoni carbonat và 20ml dung dịch amoniac TT.009 nước cất, định mức đến 100ml.
Dung dịch amoni clorrid TT.018
Dung dịch amoni clorrid 10,5% m/v (~2N) trong nước.
Dung dịch amoni clorrid/amoni hydroxid TT.019
Trộn với thể tích tương đương dung dịch amoniac đặc TT.010 và dung dịch amoni clorid bão hòa.
Dung dịch amoni citrat (PbT) TT.020
Hoà tan 40g acid citric trong 90ml nước. Thêm 2-3 giọt dung dịch phenol đỏ TT.178, sau đó thêm từ từ dung dịch amoniac đặc TT.010 cho đến khi dung dịch có màu đỏ. Loại chì bằng cách chiết nhiều lần với dung dịch dithizon (dùng để chiết) TT.096 đến khi dung dịch dithizon chiết ra có màu xanh cam.
Dung dịch amoni molybdat TT.021
Hoà tan 6,5g bột acid molybdic (85%) trong hỗn hợp 14ml nước cất và 14,5ml dung dịch amoniac đặc TT.010. Làm lạnh dung dịch, thêm từ từ dung dịch này vừa thêm vừa khuấy vào dung dịch gồm 32ml acid nitric đặc với 40ml nước cất (đã được làm lạnh trước). Để yên trong 48 giờ, lọc qua bông thủy tinh. Dung dịch sẽ bị biến chất nếu lâu không sử dụng. Trước khi sử dụng thử dung dịch bằng cách lấy 5ml dung dịch cần thử thêm 2ml dung dịch natri phosphat TT.234, nếu không tạo kết tủa màu vàng (tạo ngay hoặc sau khi khuấy nhẹ) thì dung dịch không sử dụng được. Giữ dung dịch trong chỗ tối. Nếu trong quá trình giữ trong dung dịch xuất hiện kết tủa, khi sử dụng chỉ dùng phần dung dịch trong.
Dung dịch amoni molybdat/acid sulfuric TT.022
Hoà tan 18,8g amoni molybdat trong 300ml nước cất, thêm 150ml acid sulfuric đặc, định mức đến 500ml bằng nước cất.
Dung dịch amoni oxalat TT.023
Dung dịch 3% m/v amoni oxalat [(COONH4] trong nước (~0,5N)
Dung dịch amoni sulfanilat TT.024
Thêm vào 2,5g acid sulfanilic 15ml nước cất và 3ml dung dịch amoni TT.009. Nếu cần vừa thêm vừa khuấy dung dịch amoni TT.009 cho đến khi acid tan hết. Điều chỉnh độ pH của dung dịch về 4,5 bằng dung dịch acid hydrocloric loãng TT.127, sử dụng dung dịch bromocresol xanh TT.054 làm chỉ thị màu, định mức đến 25ml bằng nước cất.
Dung dịch amoni sulfid TT.025
Bão hòa dung dịch amoni TT.009 với hydro sulfid (H2S), thêm tiếp dung dịch amoni TT.009, thể tích dung dịch amoni TT.009 thêm vào bằng 2/3 thể tích dung dịch amoni TT.009 với hydro sulfid (H2S). Cặn sau nung dung dịch không được quá 0,05%. Dung dịch không được vẩn đục khi thêm dung dịch magie sulfat TT.146, hoặc dung dịch calci clorid TT.061. Dung dịch sẽ không dùng được khi có kết tủa sulphua. Giữ dung dịch trong lọ thủy tinh màu, nhỏ, đầy dung dịch, trong chỗ tối và lạnh.
Dung dịch amoni thiocyanat TT.026
Dung dịch amonithiocyanat (NH4SCN) trong nước nồng độ 7,6% m/v (~1N)
Dung dịch amoni thiocyanat/coban nitrat TT.027
Hoà tan 17,4g amoni thiocyanat và 2,8g coban nitrat, trong nước cất, định mức đến 100ml
Dung dịch amilaza TT.028
Hoà tan 0,2g amilaza (tinh thể) trong 100ml nước cất, lắc đều và lọc. Pha dung dịch ngay trước khi sử dụng.
Dung dịch anthron TT.029
Hòa tan, khoảng 0,1g anthron trong 100ml acid sulfuric đặc. Pha dung dịch ngay trước khi sử dụng.
Dung dịch antimon (Sb) TT.030
Hòa tan 2,742g antimon kali tartrat trong nước cất, định mức đến 100ml. Lấy 5ml dung dịch này pha loãng và định mức bằng nước cất đến 500ml, 1ml dung dịch chứa 0,001mg Sb
Dung dịch antimon triclorid TT.031
Rửa bề mặt tinh thể antimon triclorid bằng clorofom khan đến khi dung dịch rửa trong. Pha dung dịch antimon triclorid bão hòa trong clorofom khan. Giữ dung dịch trong chai tránh ánh sáng, giữ lạnh, pha dung dịch ngay trước khi sử dụng.
Dung dịch asen (As) TT.032
Hút 1ml dung dịch asen đặc TT.033 pha loãng và định mức bằng nước cất đến 100ml. Pha dung dịch ngay trước khi sử dụng. 1ml dung dịch này chứa 0,01mg As.
Dung dịch asen đặc TT.033
Hòa tan 0,132g asen trioxid trong 50ml acid hydrocloric (HCl trong nước cất nồng độ 25% m/v) định mức đến 100ml
Dung dịch acid asen (III) TT.034
Hòa tan 1g acid asen (III) trong 30ml dung dịch natri hydroxid (1 trong 40), đun nóng. Để nguội thêm từ từ acid acetic băng đến 100ml
Dung dịch bari clorid TT.035
Dung dịch bari clorid (BaCl2.2H2O) trong nước nồng độ 12% m/v (~1N)
Dung dịch bari diphenilamin sulfonat TT.036
Dung dịch bari p-diphenilamin sulfonat trong nước nồng độ 0,3% m/v.
Dung dịch benzidin TT.037
Hòa tan 50mg benzidin trong 10ml acid acetic băng, định mức đến 100ml bằng nước cất, lắc đều.
Chú ý: benzidin là chất độc.
Dung dịch Bertrand A TT.038
Hòa tan 40g đồng sunfat sạch trong nước cất, định mức đến 1000ml. Giữ trong lọ thủy tinh dung tích 1000ml
Dung dịch Bertrand B TT.039
Hòa tan 200g kali natri tartrat và 150g natri hydroxid trong nước cất, định mức đến 1000ml. Giữ trong lọ thủy tinh nút cao su.
Dung dịch Bertrand C TT.040
Hòa tan 50g sắt (III) sunfat (không khử kali permanganat) trong nước cất. Thêm 200ml acid sulfuric đặc, định mức bằng nước cất đến 1000ml.
Dung dịch Bertrand D TT.041
Hòa tan 5g kali permanganat trong nước cất, định mức đến 1000ml
Chuẩn hóa: Hòa tan 0,25g amoni oxalat trong 100ml nước cất, thêm 2ml acid sulfuric đặc. Đun nóng dung dịch này đến 60 - 700C, chuẩn độ bằng dung dịch Bertrand D. Gọi thể tích dung dịch Bertrand D tiêu tốn là v(ml) thì 1ml dung dịch Bertrand D tương đương với 0,2238/v (g) Cu.
Dung dịch 2,2’-bipyridin TT.042
Hòa tan 0,1g 2,2’-bipyridin trong 50ml cồn tuyệt đối (đã tinh chế lại) TT.104
Dung dịch bismutnitrat (I) TT.043
Đun hồi lưu 5g bismut nitrat (Bi(NO3)3.5H2O) trong 7,5ml acid nitric và 10ml nước cất đến khi tan hoàn toàn. Làm nguội, lọc và định mức đến 250ml bằng nước cất.
Dung dịch bismut nitrat (II) TT.044
Hòa tan 5g bismut nitrat (Bi(NO3)3.5H2O) trong 25ml nước cất và 25ml acid acetic băng. Định mức đến 250ml bằng nước cất.
Dung dịch đệm borax TT.045
Hòa tan 3,0g borax trong 90ml nước cất. Chiết nhiều lần (mỗi lần 5ml) với dung dịch hỗn hợp 1 thể tích dung dịch diphenylthiocarbazon (PbT) và 4 thể tích clorofom, lắc liên tục cho đến khi dung dịch chiết ra có màu xanh hoặc tím. Tiếp tục chiết nhiều lần, mỗi lần 10ml clorofom cho đến khi dịch clorofom chiết ra không màu. Loại bỏ phần dịch chiết hữu cơ. Định mức dung dịch bằng nước cất đến 100ml
Dung dịch acid boric TT.046
Trong bình định mức 1000ml, hòa tan 5,0g acid boric trong 500ml nước cất. Thêm 25ml dung dịch chỉ thị trong cồn (67mg metyl đỏ và 33mg bromocresol xanh trong 100ml etanol 96%) và 200ml etanol. Định mức đến vạch bằng nước cất. 5ml dung dịch chỉ thị acid boric có màu đỏ phải chuyển màu xanh khi nhỏ tối đa 3 giọt dung dịch NaOH 0,01N vào.
Dung dịch bromid/bromat TT.047
(Khoảng 0,1N brom) (7,991g Br/l). Hòa tan 3,0g kali bromat (KBrO3) và 15g kali bromid (KBr) trong nước cất và định mức đến 1000ml.
Chuẩn hóa: Hút chính xác 25ml dung dịch này vào bình 500ml, pha loãng với 120ml nước cất. Thêm 5ml acid hydrocloric, đậy nút, lắc nhẹ. Thêm 5ml kali iodid TT.200, đậy nút, lắc đều hỗn hợp, bảo quản yên trong 5 phút và chuẩn iod tự do bằng natri thiosulfat 0,1N, thêm dung dịch hồ tinh bột TT.242 khi gần tới điểm tương đương. Tính nồng hiệu chuẩn. Giữ dung dịch trong lọ màu, nút kín.
Dung dịch brom (nước brom) TT.048
Dung dịch bão hòa brom trong nước. Chuẩn bị bằng cách lắc 2-3ml brom (Br2) trong 100ml nước lạnh, trong lọ nút nhám kín, nút được bôi mỡ. Giữ trong chỗ tối, tránh ánh sáng, lạnh.
Dung dịch brom/acid acetic TT.049
Hòa tan 5ml brom trong 145ml dung dịch kali acetat trong acid acetic TT.188. Chuẩn bị dung dịch ngay trước khi sử dụng.
Dung dịch brom/bromid TT.050
Cho 1ml brom vào 300ml acid acetic băng đã làm bão hòa bằng 5g kali bromid. Chuẩn hóa 15ml dung dịch này cần khoảng 50ml dung dịch natri thiosulfat. Giữ dung dịch trong chai thủy tinh màu và trong chỗ tối. Cần chuẩn hóa dung dịch ít nhất 1 lần/ngày khi sử dụng.
Dung dịch brom/acid acetic băng TT.051
Hòa tan 1,5g brom trong acid acetic băng, định mức đến 100ml. 1ml dung dịch này tương đương với khoảng 2ml dung dịch natri thiosulfat 0,1N.
Dung dịch brom/acid hydrocloric TT.052
Trộn 1ml dung dịch brom/kali bromid TT.053 với 100ml acid hydrocloric không có asen.
Dung dịch brom/kali bromid TT.053
Hòa tan 30g brom và 30g kali bromid trong nước cất và định mức đến 100ml
Dung dịch bromocresol xanh TT.054
Hòa tan 0,05g bromocresol xanh trong 100ml etanol, lọc nếu cần. Để làm chỉ thị cho xác định pH, hòa tan 0,05g trong 1,4ml natri hydroxid 0,05N, định mức bằng nước cất không có CO2 đến 100ml
Dung dịch bromocresol xanh/metyl đỏ TT.055
Trộn dung dịch bromocresol xanh TT.054 với dung dịch metyl đỏ TT.160 theo thể tích tương đương.
Dung dịch bromocresol tím TT.056
Hòa tan 0,25g bromocresol tím trong 20ml natri hydroxid 0,05N, định mức bằng nước cất đến 250ml
Dung dịch bromophenol xanh TT.057
Hòa tan 0,1g bromophenol xanh trong 100ml etanol loãng (1 trong 2), lọc nếu cần. Để làm chỉ thị cho xác định pH, hòa tan 0,1g trong 3,0ml natri hydroxid 0,05N, định mức bằng nước cất không có CO2 đến 200ml.
Dung dịch bromophenol xanh (cho acid citric) TT.058
Trộn dung dịch bromophenol xanh TT.057 với etanol theo tỷ lệ thể tích tương đương, điều chỉnh pH về 7,0 bằng cách thêm dung dịch natri hydroxid 0,01N.
Dung dịch bromophenol xanh/natri hydroxid TT.059
Hòa tan 0,1 bromophenol xanh trong 3ml natri hydroxid 0,05N trộn đều và định mức đến 25ml bằng nước cất.
Dung dịch bromothymol xanh TT.060
Hòa tan 0,1g bromothymol xanh trong 100ml etanol loãng (1 trong 2), lọc nếu cần. Để làm chỉ thị cho xác định pH, hòa tan trong 3,2ml natri hydroxid 0,05N, định mức bằng nước cất không có CO2 đến 200ml.
Dung dịch calci clorid TT.061
Dung dịch calci clorid (CaCl2.2H2O) trong nước nồng độ 7,5% m/v (~1N)
Dung dịch calci hydroxid TT.062
Dung dịch chứa khoảng 0,14g Ca(OH)2 trong 100ml. Pha dung dịch như sau: Cho 3,0g [Ca(OH)2] vào 1000ml nước cất, lắc liên tục trong khoảng 1 giờ. Để yên cho calci hydroxid dư lắng xuống, lọc lấy dung dịch trong phía trên.
Dung dịch Carr-Price TT.063
Lấy 01 lọ antimon triclorid 100g chưa sử dụng, kín, còn nguyên. Mở nắp và cho ngay toàn bộ animoan triclorid bên trong vào một lọ miệng rộng, nút nhám thủy tinh đã chứa sẵn khoảng 100ml clorofom. Hoặc cân antimon triclorid sau đó thêm clorofom vào với tỷ lệ 100ml clorofom/25g antimon triclorid. Hòa tan bằng cách làm ấm và lắc liên tục trong vài giờ, sau đó lọc dung dịch qua natri sulfat khan vào chai sạch, khô, có màu vàng, nút thủy tinh. Giữ dung dịch này tại nhiệt độ phòng, trong chỗ tối khi không sử dụng. Dung dịch này bền trong thời gian dài, nhưng tốt nhất chỉ nên pha đủ dùng trong thời gian 1 tháng. Rửa tất cả các dụng cụ thủy tinh tiếp xúc với dung dịch này bằng clorofom, hỗn hợp etanol/ete hoặc acid hydrocloric (loãng hoặc đặc) trước khi rửa bởi vì antimon triclorid không tan trong nước.
Dung dịch ceri amoni nitrat TT.064
Hòa tan 6,25g ceri amoni nitrat [(NH4)2Ce(NO3)6] trong 100ml acid nitric 0,25N. Dung dịch dùng trong 3 ngày kể từ lúc pha.
Dung dịch cloral hydrat TT.065
Hoà tan 50g cloral hydrat trong dung dịch hỗn hợp giữa 15ml nước cất và 10ml glycerol.
Dung dịch clo (nước clo) TT.066
Dung dịch bão hòa clo trong nước. Giữ dung dịch trong lọ nhỏ, đầy, chống ánh sáng, thậm chí trong điều kiện tránh tiếp xúc với không khí và ánh sáng, nước clo dễ bị hỏng nên phải giữ dung dịch này trong chỗ mát, tối. Tốt nhất nên chuẩn bị ngay trước khi sử dụng.
Dung dịch cromat TT.067
Hòa tan 0,0566g kali dicromat (K2Cr2O7) trong 1000ml nước cất. 1ml dung dịch này chứa 0,02mg Cr.
Dung dịch crom trioxid (dung dịch acid cromic) TT.068
Dung dịch crom trioxid trong nước nồng độ 3% m/v.
Dung dịch acid cromotropic TT.069
Trong bình định mức 1000ml, hòa tan 2,0g acid cromotropic (muối dinatri 4,5-dihydroxy-2,7-naphtalen-disulfonat) trong 40ml nước cất. Định mức đến vạch bằng dung dịch acid sulfuric 15M.
Dung dịch coban clorid TT.070
Hòa tan 2g coban clorid (CoCl2.6H2O) trong 1ml acid hydrocloric, định mức đến 100ml bằng nước cất.
Dung dịch coban uranyl acetat TT.071
Hòa tan (đun nóng nhẹ) 40g uranyl acetat [UO2(C2H3O2)2.2H2O] trong 30g acid acetat băng, định mức đến 500ml bằng nước cất. Tương tự, cân 200g coban acetat [Co(C2H3O2)2.4H2O] hoà tan trong dung dịch hỗn hợp giữa 30g acid acetic băng và nước cất, định mức bằng nước cất đến 500ml. Trộn hai dung dịch này lại trong khi vẫn còn ấm. Làm mát dung dịch đến 200C trong khoảng 2 giờ bảo quản loại muối dư ra khỏi dung dịch, sau đó lọc qua giấy lọc khô.
Dung dịch congo đỏ TT.072
Hòa tan 0,1g congo đỏ (natri diphenyl-diazo-bis-α-naphtylaminsulfonat) (C32H22N6O6S2Na2) trong 20ml etanol 20%, thêm nước định mức đến 100ml.
Dung dịch đồng sulfat TT.073
Hòa tan 34,639g CuSO4.5H2O trong nước cất, định mức đến 500ml. Lọc qua bông thủy tinh hoặc giấy lọc. Kiểm tra hàm lượng Cu trong dung dịch (tốt nhất là dùng phương pháp điện phân), điều chỉnh bảo quản có hàm lượng 440,9mg Cu/25ml.
Dung dịch cresol đỏ TT.074
Tán nhỏ 0,1g cresol đỏ trong cối có 26,2ml dung dịch natri hydroxid 0,01N đến khi tan hoàn toàn, thu dung dịch này vào bình định mức, tráng cối bằng nước cất, thu dịch tráng cối vào bình định mức, pha loãng, định mức bằng nước cất đến 250ml.
Dung dịch cresol đỏ/thymol xanh TT.075
Trộn 15ml dung dịch thymol xanh TT.251 với 5ml dung dịch cresol đỏ TT.074, lắc đều.
Dung dịch đồng acetat đặc TT.076
Hòa tan 13,3g đồng acetat trong 5ml acid acetic và 195ml nước cất.
Dung dịch đồng citrat (Thuốc thử định tính Benedict) TT.077
Dung dịch 1: Hòa tan 173g natri citrat (C6H5Na3O7.2H2O) và 117g natri carbonat (Na2CO3.2H2O) trong khoảng 700ml nước cất, lọc bằng giấy lọc nếu cần. Dung dịch 2: Hoà tan 17,3g đồng sulfat (CuSO4.5H2O) vào trong 10ml nước cất. Từ từ thêm dung dịch 2 vào dung dịch 1, vừa thêm vừa khuấy đều. Làm nguội dung dịch hỗn hợp và định mức đến 1000ml, lắc đều.
Dung dịch đồng nitrat TT.078
Dung dịch đồng nitrat [Cu(NO3).3H2O] trong nước nồng độ 2,4% m/v.
Dung dịch đồng sulfat TT.079
Dung dịch đồng sulfat [CuSO4.5H2O] trong nước nồng độ 12,5% m/v.
Dung dịch đồng sulfat/amoni TT.080
Hòa tan 0,4g đồng sulfat [CuSO4.5H2O] trong 50ml dung dịch hỗn hợp dung dịch amoni TT.009 và dung dịch citric acid (1 trong 5) theo tỷ lệ 2:3.
Dung dịch đồng tartrat kiềm (dung dịch Fehling) TT.081
Dung dịch A: hoà tan cẩn thận 34,66g đồng sulfat [CuSO4.5H2O] dạng tinh thể nhỏ, hoàn toàn không có sự lên hoa hay dính do hút ẩm, vào nước cất, định mức đến 500ml. Giữa dung dịch trong lọ vừa với thể tích dung dịch. Dung dịch B: Hòa tan 173,0g tinh thể kali natri tartrat (KNaC4H4O6.4H2O) và 50g natri hydroxid (NaOH) trong nước cất, định mức đến 500ml. Giữ dung dịch này trong lọ vừa với thể tích dung dịch và chịu ăn mòn kiềm. Ngay trước khi sử dụng trộn hai dung dịch với nhau theo tỷ lệ thể tích tương đương.
Dung dịch cyanogen bromid TT.082
Hòa tan 5,0g cyanogen bromid [CNBr] trong nước, định mức đến 50ml.
Chú ý: Pha dung dịch này trong tủ hút, vì cyanogen bromid bay hơi ngay ở điều kiện thường, khí này có tính kích thích mạnh và độc.
Dung dịch 4,4’-diaminodiphenylamin TT.083
Dung dịch bão hòa 4,4’-diaminodiphenylamin trong cồn. Tán nhỏ 4,4’-diaminodiphenylamin sulfat với lượng nhỏ etanol, thêm etanol. Chuyển dung dịch này vào bộ bình đun hồi lưu. Đun trên bể cách thủy.
Dung dịch di-β-naphtylthiocarbazon/clorofom TT.084
Cho 0,1g di-β-naphtylthiocarbazon vào trong 100ml carbon tetraclorid. Pha loãng dung dịch này với clorofom theo tỷ lệ 1:40.
Dung dịch 2,6-diclophenol-indophenol TT.085
Hòa tan 0,1g natri 2,6-diclophenol-indophenol (C12H6Cl2NNaO2) trong 100ml nước ấm, lọc dung dịch. Dung dịch dùng trong 3 ngày kể từ lúc pha.
Dung dịch 2,7-dihydronaphtalen TT.086
Hòa tan 0,1g 2,7-dihydronaphtalen trong 1000ml acid sulfuric, bảo quản yên cho đến khi dung dịch có màu vàng. Nếu dung dịch có màu sẫm thì loại bỏ và pha dung dịch khác bằng acid sulfuric khác. Giữ dung dịch này trong lọ thủy tinh màu, dung dịch bền trong 1 tháng từ lúc pha.
Dung dịch p-dimetylaminobenzal-dehyd TT.087
Hoà tan 0,125g p-dimetylaminobenzal-dehyd trong hỗn hợp (đã được làm lạnh) giữa 60ml acid sulfuric đặc và 35ml nước cất, thêm 0,05ml dung dịch sắt (III) clorid TT.112. Dung dịch sử dụng được trong 7 ngày kể từ lúc pha.
Dung dịch dimetylglyoxim TT.088
Dung dịch dimetylglyoxim trong etanol nồng độ 1% m/v.
Dung dịch 2,4-dinitrophenylhydrazin TT.089
Hòa tan 0,2g 2,4-dinitrophenylhydrazin trong 100ml acid sulfuric 85%. Lọc qua phễu lọc chịu acid nếu cần. Giữ dung dịch trong chai thủy tinh màu, ở chỗ tối, giữ lạnh. Dung dịch sử dụng trong 2 tuần kể từ lúc pha.
Dung dịch diphenylamin TT.090
Dung dịch diphenylamin [(C6H5)2NH] trong acid sulfuric nồng độ 1% m/v. Dung dịch phải trong, không màu.
Dung dịch diphenylcarbazid TT.091
Hòa tan 0,125g diphenylcarbazid [(C6H5NHNH)2CO] trong hỗn hợp 25ml aceton và 25ml nước cất. Pha dung dịch ngay trước khi sử dụng.
Dung dịch diphenylcarbazon TT.092
Dung dịch diphenylcarbazon [(C13H12N4O] trong etanol nồng độ 1% m/v. Giữ dung dịch trong chai thủy tinh màu.
Dung dịch diphenylthiocarbazon (PbT) TT.093
Chiết 15ml dung dịch diphenyl-thiocarbazon [(C6H4N:NCSNH-NHC6H5] trong clorofom nồng độ 0,1% m/v bằng hỗn hợp giữa 5ml dung dịch amoniac TT.009 và 50ml nước cất, tiến hành chiết 2 lần. Gộp 2 phần dịch chiết pha nước, acid hoá bằng dung dịch acid hydrocloric loãng (PbT) TT.128. Chiết bằng 100ml clorofom, rửa dịch chiết clorofom bằng nước cất, 2 lần X 10ml, lọc pha phễu lọc khô. Kiểm tra dung dịch này theo phương pháp xác định kẽm (xem chuyên luận Titan dioxid), sử dụng 5ml dung dịch kẽm sulfat chuẩn TT.262 định mức đến 25ml dung dịch acid trong quy trình phân tích. Pha loãng với clorofom bảo quản 3ml dung dịch này tương đương với 1ml dung dịch kẽm sulfat chuẩn TT.262. Pha dung dịch này ngay trước khi sử dụng.
Dung dịch α,α-dipyridin TT.094
Dung dịch α,α-dipyridin [(C10H8N2] trong etanol tuyệt đối nồng độ 0,2% m/v.
Dung dịch dithizon TT.095
Hòa tan 25,6g dithizon trong 100ml etanol.
Dung dịch dithizon chiết TT.096
Hòa tan 30mg dithizon trong 1000ml cloroform và thêm 5ml etanol. Bảo quản dung dịch trong tủ lạnh. Trước khi sử dụng, trộn hỗn hợp trên với acid nitric 1% với tỷ lệ thể tích 2:1, lắc đều, loại bỏ lớp acid nitric. Dung dịch thu được có thể sử dụng trong 1 tháng kể từ lúc pha.
Dung dịch dithizon chuẩn TT.097
Hòa tan 10mg dithizon trong 1000ml cloroform, đựng dung dịch trong lọ sạch có nút thủy tinh. Bọc kín bảo quản tránh ánh sáng và giữ trong tủ lạnh.
Dung dịch Dragendorff TT.098
Dung dịch 1: Cân 0,85g bismut nitrat bazơ, hòa tan trong 10ml acid acetic và 40ml nước cất.
Dung dịch 2: Cân 8,0g kali iodid, hoà tan trong 20ml nước cất.
Trộn 5ml dung dịch 1; 5ml dung dịch 2; 20ml acid acetic và 100ml nước cất. Pha dung dịch ngay trước khi sử dụng.
Dung dịch chỉ thị hấp thụ Eosin Y TT.099
Dung dịch Eosin Y trong nước nồng độ 0,5%.
Dung dịch ericrom đen TT.100
Hòa tan 0,22g ericrom đen T và 2g hydroxylamin hydroclorid (NH2OH.HCl) trong metanol, định mức đến 50ml, lọc. Giữ dung dịch trong chai thủy tinh màu, tránh ánh sáng, dung dịch dùng trong 2 tuần sau khi pha.
Etanol (cồn) TT.101
Dung dịch chứa 95% etanol v/v.
Etanol tuyệt đối (cồn tuyệt đối) TT.102
Dung dịch chứa 99,5% etanol v/v.
Etanol không có aldehyd TT.103
Cho vào 1000ml cồn, thêm 5ml acid sulfuric đặc và 20ml nước cất, tiến hành chưng cất. Thêm 10g bạc nitrat và 1g kali hydroxid vào 1000ml dung dịch chưng cất thu được. Đun hồi lưu trong 3 giờ, sau đó cất thu etanol không có aldehyd.
Etanol tuyệt đối đã tinh chế TT.104
Cho vào cồn tuyệt đối kali permaganat (khoảng 0,1%) và kali hydroxid (khoảng 0,1%), cất bằng bộ chưng cất toàn bộ bằng thủy tinh.
Etanol 90% TT.105
(tại 15,560) pha loãng 94,8ml etanol bằng nước cất định mức đến 100ml tại 250C.
Etanol 80% TT.106
(tại 15,560) pha loãng 84,3ml etanol bằng nước cất định mức đến 100ml tại 250C.
Etanol 72% TT.107
Trộn 360ml etanol tuyệt đối đã tinh chế TT.104 với 150ml nước cất.
Etanol 70% TT.108
(tại 15,560) pha loãng 73,7ml etanol bằng nước cất định mức đến 100ml tại 250C.
Dung dịch p-etoxycrysoidin TT.109
Hoà tan 50mg p-etoxycresoidin monohydroclorid trong hỗn hợp 25ml nước và 25ml etanol, thêm 3 giọt acid hydrocloric, khuấy đều, lọc lấy dung dịch trong nếu cần.
Dung dịch sắt (III) amoni sulfat TT.110
Dung dịch sắt (III) amoni sulfat [FeNH4(SO4)2.2H2O] trong nước nồng độ 8% m/v.
Dung dịch sắt (III) amoni sulfat/acid hydroclorid TT.111
Dung dịch sắt (III) amoni sulfat [FeNH4(SO4)2.2H2O] trong acid hydrocloric nồng độ 0,1% m/v.
Dung dịch sắt (III) clorid TT.112
Dung dịch sắt (III) clorid [FeCl3.6H2O] trong nước nồng độ 9% m/v (~1N).
Dung dịch sắt (III) clorid trong etanol TT.113
Dung dịch sắt (III) clorid [FeCl3.6H2O] trong etanol tuyệt đối nồng độ 0,2% m/v. Pha dung dịch ngay trước khi sử dụng.
Dung dịch sắt (III) clorid loãng TT.114
Pha loãng dung dịch sắt (III) clorid TT.112 trong nước, định mức đến 100ml. Pha dung dịch ngay trước khi sử dụng.
Dung dịch sắt (III) clorid/acid hydrocloric TT.115
Hòa tan 5g sắt (III) clorid [FeCl3.6H2O] trong 5ml acid hydrocloric, định mức bằng nước cất đến 100ml.
Dung dịch sắt (III) sulfat TT.116
Cho 50g sắt (III) sulfat vào 500ml nước cất, lắc đều. Thêm vào dung dịch này 200ml acid sulfuric, lắc kỹ cho tan hoàn toàn, thêm nước cất, định mức đến 1000ml
Dung dịch sắt (III) sulfat acid TT.117
Trộn 7,5ml acid sulfuric vào 100ml nước cất, lắc đều. Hòa tan 80g sắt (II) sulfat vào dung dịch này, đun nóng cho tan hoàn toàn. Trộn 7,5ml acid nitric vào 20ml nước cất, đun nóng nhẹ. Trộn dung dịch này vào dung dịch sắt (II) sulfat lắc kỹ cho tan hoàn toàn, thêm nước cất, định mức đến 1000ml. Cô dung dịch hỗn hợp đến khi màu của dung dịch chuyển từ đen sang đỏ. Kiểm tra sự có mặt của sắt (II) trong dung dịch, nếu có thêm vài giọt acid nitric và đun tiếp. Để nguội dung dịch thêm nước cất đến đủ 110ml.
Dung dịch sắt (II) sulfat TT.118
Hoà tan 8,0g tinh thể sắt (II) sulfat sạch [FeSO4.7H2O] vào khoảng 100ml nước cất vừa đun sôi bảo quản nguội. Pha dung dịch ngay trước khi sử dụng.
Dung dịch sắt (II) sulfat acid TT.119
Hoà tan 7,0g tinh thể sắt (II) sulfat sạch [FeSO4.7H2O] vào khoảng 90ml nước cất vừa đun sôi bảo quản nguội. Thêm acid sulfuric đến đủ 100ml (nồng độ khoảng 0,25N). Thường xuyên chuẩn lại dung dịch bằng kali permanganat 0,25N.
Dung dịch fluorescen TT.120
Dung dịch natri fluorescen trong etanol 50% nồng độ 0,1% m/v.
Dung dịch Folin-Ciocanteu TT.121
Cho vào bình cầu cất dung tích 150ml; 10g natri tungstat (Na2WO4.2H2O), 2,5g natri molybdat (Na2MoO4.2H2O), 75ml nước cất, 5ml acid phosphoric và 10ml acid hydrocloric. Đun hồi lưu hỗn hợp này (đun đủ sôi) trong 10 giờ. Thêm 15g liti sulfat (Li2SO4.H2O), 50ml nước cất, vài giọt brom. Đun sôi hỗn hợp (không lắp sinh hàn) trong 15 phút cho đến khi thấy hơi brom thoát ra. Làm nguội hỗn hợp, định mức bằng nước cất đến 100ml, lọc. Dung dịch lọc không được có màu xanh. Trước khi dùng pha loãng dung dịch này với nước theo tỷ lệ 1:1 về thể tích.
Dung dịch formaldehyd TT.122
Dung dịch chứa ~37,0% HCHO. Dung dịch có thể chứa metanol bảo quản tránh cao phân tử hóa.
Dung dịch formalin/acidsulfuric TT.123
Trộn 0,2ml dung dịch formaldehyd TT.122 với 10ml acid sulfuric. Pha dung dịch ngay trước khi sử dụng.
Dung dịch fuchsin/acid sulfurơ TT.124
Hoà tan 0,5g fuchsin bazơ trong 300ml nước cất, làm nguội. Thêm vào dung dịch này 50ml dung dịch chứa 5g natri sulfit khan trong 50ml nước cất, thêm 5ml acid hydrocloric, lắc đều. Định mức dung dịch đến 500ml, bảo quản yên trong 5 giờ. Bảo quản trong lọ thủy tinh màu, tránh ánh sáng, bảo quản lạnh.
Dung dịch acid hydriodic TT.125
Cất acid hydriodic qua phospho đỏ, tạo môi trường khí CO2 trong bộ cất trong suốt quá trình cất. Nhiệt độ cất luôn đảm bảo 1260-1270C, dung dịch cất phải trong, không màu. Bảo quản trong lọ thủy tinh màu, nút thủy tinh đã được đuổi hết không khí bằng khí CO2, nút được gắn kín bằng parafin. Giữ trong chỗ tối, mát.
Dung dịch acid hydrocloric brom hóa TT.126
Trộn 1ml dung dịch brom với 100ml acid hydrocloric.
Dung dịch acid hydrocloric loãng TT.127
Dung dịch chứa HCl 10% m/v trong nước. Pha loãng 266ml dung dịch acid hydrocloric 36% với nước cất, định mức đến 1000ml
Dung dịch acid hydrocloric loãng (PbT) TT.128
Dung dịch chứa HCl 10% m/v trong nước. Đạt kiểm tra như sau: Lấy 10ml dung dịch amoniac loãng (PbT) TT.011 thêm 1ml kali cyanid (PbT) TT.193 định mức đến 50ml băng nước cất, thêm 2 giọt dung dịch natri sunfit 10% trong nước. Dung dịch không được sẫm màu lại.
Dung dịch thiếc clorid trong acid hydrocloric TT.129
Trộn 1ml dung dịch thiếc clorid TT.241 với 100ml dung dịch acid hydrocloric trong nước nồng độ 25% m/v.
Dung dịch hydro peroxid TT.130
Dung dịch chứa khoảng 2,5 - 3,5g H2O2 trong 100ml nước. Dung dịch có thêm chất bảo quản thích hợp nhưng nồng độ không vượt quá 0,05%.
Dung dịch hydro sulfid TT.131
Dung dịch bão hòa hydro sulfid trong nước. Pha bằng cách sục khí H2S đi qua nước cất lạnh. Bảo quản trong lọ thủy tinh màu, dung tích phù hợp với thể tích dung dịch. Dung dịch dùng được phải có mùi H2S mạnh và phải tạo kết tủa sunfua khi thêm thể tích tương đương dung dịch sắt (III) clorid TT.112. Bảo quản lạnh trong chỗ tối.
Dung dịch hydroxylamin hydroclorid TT.132
Hòa tan 20g hydroxylamin hydroclorid (HONH2.HCl) trong nước để thu được khoảng 65ml dung dịch, cho dung dịch này vào phễu chiết, thêm vài giọt chỉ thị pH thymol xanh, thêm dung dịch amoniac đặc TT.010 đến khi dung dịch có màu vàng, thêm 10ml dung dịch natri dietyl dithiocarbamat 4%, trộn đều, để yên 5 phút. Chiết dung dịch này nhiều lần với clorofom, mỗi lần 10-15ml, cho đến khi thử dịch clorofom chiết ra bằng cách lấy 5ml lắc với dung dịch đồng sulfat loãng không có màu vàng. Thêm dung dịch acid hydrocloric loãng (PbT) TT.128 đến khi dung dịch có màu hồng. Pha loãng và định mức bằng nước cất đến 100ml
Dung dịch 8-hydroxyquinolin TT.133
Dung dịch 8-hydroxyquinolin (oxin) trong nước nồng độ 5%.
Dung dịch Indigo Carmin (dung dịch indigotindisulfonat) TT.134
Hòa tan khối lượng Indigo Carmin (natri indigotindsulfonat) tương đương với 0,18g C18H8O8N2S2Na2 trong nước cất đến đủ 100ml. Dung dịch này dùng trong 60 ngày kể từ khi pha.
Dung dịch iod TT.135
Hoà tan 14g iod vào dung dịch chứa 36g kali iodid trong 100ml nước, thêm 3 giọt acid hydrocloric, định mức đến 100ml bằng nước cất.
Dung dịch chỉ thị sắt TT.136
Cho 62,5ml sắt (III) amoni sulfat vào lọ dung tích 1000ml, hoà tan trong 500ml nước cất, thêm 450ml acid nitric đặc, trộn đều.
Dung dịch sắt chuẩn (0,01mg Fe/ml) TT.137
Hòa tan 0,70g Fe(NH4)2(SO4)2.6H2O vào 50ml nước cất, thêm 20ml dung dịch H2SO4 loãng (1:15). Định mức đến 1000ml bằng nước cất, lắc đều. Lấy 10ml dung dịch này định mức đến 100ml
Dung dịch chì chuẩn (0,01mg Pb/ml) TT.138
Hòa tan 0,1598g chì nitrat [Pb(NO3)2] vào nước cất, thêm 1ml HNO3. Định mức đến 1000ml. Lấy 10ml dung dịch này định mức đến 100ml. Pha dung dịch ngay trước khi sử dụng.
Dung dịch chì acetat TT.139
Hòa tan 9,5g tinh thể trong, sạch chì acetat [Pb(CH3COO)2.2H2O] vào nước cất sôi, định mức đến 100ml. Bảo quản dung dịch trong lọ kín.
Dung dịch chì acetat bazơ TT.140
Trộn 10 phần bột chì oxid (PbO), 30 phần chì acetat [Pb(CH3COO)2.2H2O] và 5 phần nước cất, đun nhẹ hỗn hợp, thỉnh thoảng lắc đều, cho đến khi hỗn hợp có màu trắng. Thêm 95 phần nước, đun trong 1 giờ, thỉnh thoảng lắc đều, để nguội, lọc. Thêm nước nếu cần để thu được dung dịch có trọng lượng riêng 1,225-1,230
Dung dịch chì subacetat TT.141
Tán nhỏ 14g chì monoxid (PbO), 10ml nước cất thành dạng bột nhão, chuyển hỗn hợp thu được vào lọ thủy tinh, rửa cối bằng 10ml thu nước rửa vào lọ. Hòa tan 22g [Pb(CH3COO)2.2H2O] trong 70ml nước cất, cho hỗn hợp này vào lọc chứa chì monoxid, lắc đều trong 5 phút. Sau đó để trong 7 ngày, thỉnh thoảng lắc đều. Lọc, thêm nước cất vừa đun sôi vào dung dịch lọc tới đủ 100ml
Dung dịch chì subacetat loãng TT.142
Pha loãng 3,25ml dung dịch chì subacetat TT.141 với nước cất vừa đun sôi tới đủ 100ml. Bảo quản trong lọ nhỏ dung tích phù hợp với thể tích dung dịch.
Dung dịch quỳ TT.143
Đun 10g thuốc thử quỳ tinh khiết với 40ml etanol 90% trong 1 giờ, loại bỏ phần dung dịch trong, tiếp tục đun phần cặn với 30ml etanol 90%, loại phần dung dịch cồn. Đun phần cặn với 100ml nước cất trong 1 giờ, để nguội và lọc.
Dung dịch mangan sulfat TT.144
Hòa tan 90g mangan sulfat trong 200ml nước cất, 175ml acid phosphoric và 350ml dung dịch acid sulfuric (1 trong 2). Thêm nước cất đến đủ 1000ml.
Dung dịch hỗn hợp magnesia TT.145
Hòa tan 5,5g magie clorid (MgCl2.6H2O) và 7g amoni clorid (NH4Cl) trong 65ml nước cất, để dung dịch trong lọ kín vài ngày. Nếu dung dịch đục thì lọc trước khi sử dụng.
Dung dịch magie sulfat TT.146
Hòa tan 12g tinh thể magie sulfat (MgSO4.7H2O), chọn từ sự tạo hoa tinh thể, trong nước cất đến đủ 100ml dung dịch.
Dung dịch Malachit xanh TT.147
Dung dịch malachit xanh oxalat trong acid acetic băng nồng độ 1% m/v
Dung dịch thuỷ ngân (II) acetat TT.148
Dung dịch thủy ngân acetat [Hg(COOCH3)2] trong acid acetic băng nồng độ 6% m/v. Bảo quản trong lọ thủy tinh màu, tránh ánh sáng.
Dung dịch thủy ngân (II) clorid TT.149
Dung dịch thủy ngân clorid [HgCl2] trong nước nồng độ 6,5% m/v (~0,5N).
Dung dịch thủy ngân (II)-kali iodid (Dung dịch Mayer) TT.150
Hòa tan 1,358g thuỷ ngân clorid [HgCl2] trong 60ml nước cất. Hòa tan 5g iodid (KI) trong 10ml nước cất. Trộn hai dung dịch này, thêm nước đến đủ 100ml.
Dung dịch thủy ngân (II)-kali iodid kiềm (Dung dịch Nessler) TT.151
Hòa tan 10g kali iodid (KI) trong 10ml nước cất. Thêm từ từ, vừa thêm vừa khuấy đều dung dịch thủy ngân clorid bão hòa trong nước cho đến khi bắt đầu tạo kết tủa bền màu đỏ. Thêm vào dung dịch này 60ml dung dịch chứa 30g kali hydroxid (30g KOH/60ml H2O) đã làm lạnh đến đông đá, sau đó thêm 1ml thủy ngân clorid bão hòa. Định mức đến 200ml bằng nước cất, để yên cho kết tủa lắng xuống, lọc lấy phần dung dịch trong. Kiểm tra: lấy ngay khi lấy 2ml dung dịch này cho vào bình chứa 100ml dung dịch amoni clorid trong nước không có amoniac (1 trong 300.000), phải tạo màu vàng nâu.
Dung dịch thuỷ ngân (II) nitrat TT.152
Hòa tan 40g thủy ngân oxid vàng (HgO) vào hỗn hợp 32ml acid nitric và 15ml nước cất (nồng độ ~ 4N). Bảo quản dung dịch trong lọ kín tránh ánh sáng.
Dung dịch thủy ngân (II) sulfat (dung dịch Denige) TT.153
Hòa tan 5g thủy ngân oxid vàng (HgO) vào 40ml nước cất, khuấy đều, trong khi khuấy cho thêm từ từ 20ml acid sulfuric, thêm 40ml nước cất. Khuấy đều cho tan hết (nồng độ ~0,5N).
Dung dịch thủy ngân (I) nitrat TT.154
Hòa tan 200g thủy ngân trong acid nitric, thêm nước cất đến đủ 1000ml. Dung dịch bảo quản trong lọ có chứa 1 ít thủy ngân kim loại.
Dung dịch p-metylaminophenol TT.155
Hòa tan 2,0g p-metylaminophenol sulfat [(HO.C6H4.NHCH3)2.H2SO4] trong 100ml nước cất. Lấy 100ml dung dịch này thêm 90ml nước cất và 20g natri bisulfit. Kiểm tra dung dịch: Lấy 4 ống nghiệm lần lượt cho vào mỗi ống 25ml dung dịch acid sulfuric 0,5N; 1ml dung dịch amoni molybdat TT.021; 1ml dung dịch vừa chuẩn bị. Thêm 5μg PO43-, 10μg PO43-, 20μg PO43- lần lượt vào ống số 1, 2, 3 tương ứng (dùng 0,5ml, 1,0ml, 2,0ml dung dịch chuẩn phosphat), để yên trong 2 giờ. 3 ống này sẽ có màu xanh da trời, cường độ màu tỷ lệ thuận với lượng phosphat thêm vào. Màu của ống chứa 5μg PO43- đậm hơn rõ ràng so với ống trắng.
Dung dịch metylen xanh TT.156
Hòa tan 0,125g metylen xanh trong 100ml etanol, định mức đến 250ml bằng etanol
Dung dịch metylen xanh loãng TT.157
Pha loãng 1ml dung dịch metylen xanh TT.157 trong nước tới đủ 100ml
Dung dịch metyl da cam TT.158
Hòa tan 0,1g metyl da cam trong 100ml nước cất, lọc nếu cần.
Dung dịch metyl da cam/xylencyanol FF TT.159
Hòa tan 1,0g metyl da cam và 1,4g xylencyanol FF trong 500ml etanol 50% v/v.
Dung dịch metyl đỏ TT.160
Hòa tan 0,1g metyl đỏ trong 100ml etanol, lọc nếu cần. Để làm chỉ thị xác định pH hòa tan 0,1g trong 7,4ml dung dịch natri hydroxid 0,05N, pha loãng bằng nước cất không có carbon dioxid, định mức đến 200ml.
Dung dịch metyl đỏ/metylen xanh TT.161
Trộn 10ml metyl đỏ TT.160 và 10ml dung dịch metylen xanh TT.156, trộn đều.
Dung dịch Millon TT.162
Cho 2ml thuỷ ngân và 20ml acid nitric vào trong bình nón, lắc đều (tiến hành trong tủ hút). Sau 10 phút cho thêm 35ml nước cất, nếu có kết tủa hoặc tinh thể xuất hiện thêm dung dịch acid nitric (1 trong 5, pha từ acid nitric đã loại oxid bằng cách sục không khí qua đến khi dung dịch acid không màu) để hòa tan hoàn toàn các chất rắn. Thêm nhỏ giọt dung dịch natri hydroxid (1 trong 10) vào dung dịch, trộn đều, đến khi dung dịch xuất hiện kết tủa dạng huyền phù. Thêm 5ml dung dịch acid nitric loãng, trộn đều. Pha dung dịch ngay trước khi sử dụng.
Chỉ thị Murexid TT.163
Thêm 0,4g murexid vào 40g bột kali sulfat, đồng nhất hóa hỗn hợp trong cối thủy tinh. Viên chứa 0,4mg murexid phối chế với kali sulfat, kali clorid có bán sẵn trên thị trường).
Dung dịch naphtalendiol TT.164
Hòa tan 0,1g 2,7-dihydoxynaphtalen trong 1000ml acid sulfuric, để yên dung dịch trong chỗ tối cho đến khi dung dịch mất màu vàng (tối thiểu 18 giờ).
Dung dịch 1-naphtol TT.165
Hòa tan 1,0g 1-naphtol trong 25ml metanol. Pha dung dịch ngay trước khi sử dụng.
Dung dịch naphtol xanh TT.166
Dung dịch naphtol xanh trong nước nồng độ 0,05% m/v.
Dung dịch α-naphtolbenzein TT.167
Dung dịch α-naphtolbenzein trong benzol nồng độ 1% m/v.
Dung dịch neutral đỏ TT.168
Dung dịch neutral đỏ trong etanol (50%) nồng độ 0,1% m/v.
Dung dịch ninhydrin (dung dịch triketonhydrinden hydrat) TT.169
Dung dịch ninhydrin (C9H4O3.H2O) trong nước nồng độ 0,2% m/v. Pha dung dịch ngay trước khi sử dụng.
Dung dịch acid nitric loãng TT.170
Dung dịch chứa khoảng 10% HNO3 m/v. Pha loãng 105ml acid nitric 70% với nước cất, định mức đến 1000ml.
Dung dịch acid nitric/acid sulfuric TT.171
Pha loãng 420ml acid nitric 70% với 580ml nước cất (thu được dung dịch HNO3 32-35% m/v), thêm 30ml acid sulfuric, lắc đều.
Dung dịch chuẩn nitrit TT.172
Hoà tan 1,5g natri nitrit (NaNO2) trong 1000ml nước không có carbon dioxid và amoniac. 1ml dung dịch chứa 1mg NO2-.
Dung dịch o-nitrobenzaldehyd TT.173
Dung dịch bão hòa o-nitrobenzaldehyd (NO2C6H4CHO) trong dung dịch natri hydroxid 2N.
Dung dịch orthophenanthrolin TT.174
Hòa tan orthophenanthrolin (C12H8N2.H2O) trong 10ml dung dịch sắt (II) sulfat (Hòa tan 1,48g tinh thể sắt (II) sulfat trong (FeSO4.7H2O) vào 100ml nước, pha dung dịch sắt (II) sulfat ngay trước khi thêm orthophenanthrolin). Bảo quản dung dịch trong lọ nút kín.
Dung dịch acid oxalic TT.175
Dung dịch acid oxalic (C2H2O4.2H2O) trong nước nồng độ 6,3% (~1N)
Dung dịch acid oxalic/acid sulfuric TT.176
Trộn acid sulfuric vào nước cất với tỷ lệ thể tích tương đương, làm nguội dung dịch. Thêm 25g acid oxalic vào 500ml dung dịch này, lắc đều.
Dung dịch 8-oxyquinolin TT.177
Hòa tan 2g 8-oxyquinolin trong 6ml acid acetic băng, thêm nước cất đến đủ 100ml. Pha dung dịch ngay trước khi sử dụng.
Dung dịch phenol đỏ (dung dịch phenolsulfonphtalein) TT.178
Hoà tan 0,1g phenolsulfonphtalein trong 100ml etanol, lọc nếu cần. Thêm nước cất đến đủ 100ml. Pha dung dịch ngay trước khi sử dụng. Để làm chỉ thị xác định pH hòa tan 0,1g trong 5,7ml dung dịch natri hydroxid 0,05N, pha loãng bằng nước cất không có carbon dioxid, định mức đến 200ml
Dung dịch phenolphtalein TT.179
Hòa tan 0,2g phenolphtalein (C20H14O4) trong 60ml etanol 90%, thêm nước đến đủ 100ml
Dung dịch phenolphtalein/thymol xanh TT.180
Hòa tan 2,0g phenolphtalein (C20H14O4) và 0,1g thymol xanh trong 100ml etanol tuyệt đối, lọc nếu cần. Pha dung dịch ngay trước khi sử dụng.
Dung dịch phenylhydrazinhydroclorid/natri acetat TT.181
Hòa tan 0,5g phenylhydrazinhydroclorid trong 10ml dung dịch natri acetat TT.218, lọc nếu cần. Pha dung dịch ngay trước khi sử dụng.
Dung dịch p-phenyl phenol TT.182
Hòa tan 0,75g p-phenyl phenol trong 50ml dung dịch natri hydroxid TT.230
Dung dịch phlorogluxin/acid hydrocloric TT.183
Hòa tan 0,1g phlorogluxin trong 1ml etanol, thêm 9ml acid hydrocloric, trộn đều. Bảo quản trong chỗ tối.
Dung dịch acid phosphomolybpdic TT.184
Hòa tan 5,0g acid phosphomolybpdic (20MoO3.2H3PO3.48H2O) nước cất. Lọc và định mức bằng nước cất đến 100ml
Dung dịch acid phosphotungstic TT.185
Dung dịch acid phosphotungstic (~24WO3.2H3PO3.48H2O) trong nước nồng độ 1% m/v.
Dung dịch platin clorid TT.186
Dung dịch platin clorid trong nước nồng độ 13% m/v (~0,5N)
Dung dịch kali acetat TT.187
Dung dịch kali acetat [KCH3COO] trong nước nồng độ 10% m/v.
Dung dịch kali acetat trong acid acetic TT.188
Hòa tan 10g kali acetat trong 100ml dung dịch hỗn hợp 90ml acid acetic băng và 10ml anhydrid acetic.
Dung dịch kali bromat/kali bromid TT.189
Hòa tan 1,4g kali bromat và 8,1g kali bromid vào nước cất, định mức đến đủ 100ml
Dung dịch kali clorid trong acid hydrocloric TT.190
Hòa tan 25g kali clorid trong 0,85ml acid hydrocloric băng và 75ml nước cất.
Dung dịch kali cromat TT.191
Dung dịch kali cromat (K2CrO4) trong nước nồng độ 10%.
Dung dịch kali cyanat TT.192
Hòa tan 1,0g kali cyanat trong 9ml nước cất. Pha dung dịch ngay trước khi sử dụng.
Dung dịch kali cyanid (PbT) TT.193
Hòa tan 50g kali cyanid trong nước tinh khiết để thu được 100ml dung dịch. Loại chì bằng cách lắc nhiều lần với dung dịch dithizon chiết TT.096, một phần dithizon khó tách ra khỏi pha nước, nếu cần loại bằng cách chiết với clorofom. Sau đó pha loãng để có dung dịch kali cyanid nồng độ 10g/100ml
Dung dịch kali dicromat (dung dịch kali bicromat) TT.194
Dung dịch kali dicromat (K2Cr2O7) trong nước nồng độ 7,5%.
Dung dịch kali fericyanid TT.195
Hoà tan 1g kali ferricyanid [K3Fe(CN)6] trong 10ml nước cất. Pha dung dịch ngay trước khi sử dụng.
Dung dịch kali ferocyanid TT.196
Hòa tan 1g kali ferrocyanid [K4Fe(CN)6.3H2O] trong 10ml nước cất. Pha dung dịch ngay trước khi sử dụng.
Dung dịch kali hydroxid TT.197
Dung dịch kali hydroxid (KOH) trong nước nồng độ 6,5% m/v (~1N)
Dung dịch kali hydroxid trong etanol TT.198
Cho 5-10g kali hydroxid (KOH) vào bình cầu cất 2 lít, thêm 1-1,5l etanol 95% đun hồi lưu trên bếp cách thủy từ 30-60 phút. Tiến hành cất thu etanol. Hòa tan 40g kali hydroxid (loại ít carbonat) trong 100ml etanol cất lại, etanol này được giữ tại 15,50C, trong khi kali hydroxid hòa tan, dung dịch phải trong.
Dung dịch kali iodat TT.199
Dung dịch kali iodat trong nước nồng độ 0,71% m/v. Bảo quản trong chỗ tối.
Dung dịch kali iodid TT.200
Dung dịch kali iodid (KI) trong nước nồng độ 16,5% m/v (~1N). Bảo quản tránh ánh sáng.
Dung dịch kali permanganat TT.201
Dung dịch kali permanganat (KMnO4) trong nước nồng độ 1% m/v.
Dung dịch kali permanganat/acid phosphoric TT.202
Hòa tan 15g kali permanganat trong dung dịch acid phosphoric (75ml acid phosphoric trong 500ml nước cất)
Dung dịch kali pyroantimonat TT.203
Cho 2g kali pyroantimonat vào 100ml nước cất đun sôi dung dịch trong 5 phút, làm nguội nhanh và thêm 10ml dung dịch kali hydroxid 15%. Để yên dung dịch trong 1 ngày, lọc.
Dung dịch kali natri tartrat TT.204
Dung dịch kali natri tartrat (KNaC4H4O6.4H2O) trong nước nồng độ 14,1% m/v.
Dung dịch kali sulfat TT.205
Dung dịch kali sulfat (K2SO4) trong nước nồng độ 1% m/v.
Dung dịch pyridin/anhydrid acetic TT.206
Trộn pyridin với anhydrid acetic theo tỷ lệ thể tích 3:1. Pha dung dịch ngay trước khi sử dụng.
Dung dịch pyridin clorid/clorofom TT.207
Cho 75g pyridin khan (C5H5N) và khoảng 400ml clorofom và ống đong dung tích 2000ml có chia độ. Cân ống đong và làm lạnh trong nước đá. Sục khí hydro clorid qua dung dịch (tốc độ chậm). Sau vài phút ngừng sụt khí, lấy ống đong ra khỏi bể nước đá, làm khô, cân, để kiểm tra lượng khí đã hấp tụ vào dung dịch. Khi lượng khí hấp thụ đạt khoảng 35g, ngừng sục khí. Làm ấm dung dịch đến nhiệt độ phòng. Đuổi hơi ra khỏi ống bằng đong không khí khô, thêm 100ml pyridin khan và định mức đến 1000ml bằng clorofom. Loại bỏ lượng dung dịch còn lại 100ml trong bình sau khi dùng.
Dung dịch Quimociac TT.208
Hòa tan 70g natri molybdat (Na2MoO4.2H2O) trong 150ml nước cất (dung dịch A). Hòa tan 60g acid citric vào hỗn hợp 85ml acid nitric và 150ml nước cất, làm nguội dung dịch (dung dịch B). Từ từ trộn dung dịch A vào dung dịch B, khuấy đều (dung dịch C). Hòa tan 5ml quinolin tổng hợp vào hỗn hợp 35ml acid nitric và 100ml nước cất (dung dịch D). Từ từ trộn đều dung dịch C và dung dịch D, để qua đêm. Lọc dung dịch, thêm 280ml aceton vào dịch lọc, định mức đến 1000ml bằng nước cất, lắc đều. Bảo quản trong lọ polyetylen.
Chú ý: dung dịch này có chứa aceton, không sử dụng gần ngọn lửa. Khi sử dụng cần đun nóng phải tiến hành trong tủ hút.
Dung dịch quinaldin TT.209
Dung dịch quinaldin đỏ trong acid acetic băng nồng độ 0,1% m/v.
Dung dịch salicylaldehyd TT.210
Dung dịch salicylaldehyd trong etanol băng nồng độ 20% m/v.
Dung dịch thuốc thử Schiff TT.211
Hòa tan 0,125g tinh thể rosalin clohydrat (C20H20CIN3) trong 1000ml nước cất, loại màu bằng acid sulfurơ.
Dung dịch thuốc thử Schiff cải tiến TT.212
Hòa tan 0,2g tinh thể rosalin clohydrat (C20H20CIN3) trong 120ml nước cất nóng. Làm nguội dung dịch, thêm 2g natri bisulfit (NaHSO3), 2ml acid hydrocloric. Định mức tới 200ml bằng nước cất. Bảo quản trong lọ thủy tinh màu tại 150C hoặc thấp hơn.
Dung dịch acid silicotungstic TT.213
Hòa tan 10,0g acid silicotungstic (SiO2.12WO3.26H2O) trong nước cất, trung hòa với dung dịch natri hydroxid 10%, sử dụng chỉ thị metyl đỏ. Pha loãng đến 100ml.
Dung dịch bạc amoni nitrat (dung dịch bạc amonionitrat) TT.214
Hòa tan 1,0 bạc nitrat trong 20ml nước cất. Thêm (nhỏ giọt) dung dịch amoniac TT.009 khuấy liên tục, cho đến khi xuất hiện kết tủa, lọc. Bảo quản dung dịch trong lọ thủy tinh màu tránh ánh sáng.
Dung dịch bạc nitrat TT.215
Dung dịch bạc nitrat (AgNO3) trong nước nồng độ 4,2% m/v (~0,25N)
Dung dịch bạc nitrat acid TT.216
Hoà tan 15g bạc nitrat trong 50ml nước cất, thêm 400ml etanol và vài giọt acid nitric đặc. Chuẩn hóa dung dịch với dung dịch amoni thiocyanat 0,05N theo phương pháp Volhard. Dung dịch này rất bền.
Dung dịch phun sương bạc nitrat TT.217
Dung dịch A: Hòa tan 50g bạc nitrat trong 450ml nước cất. Bảo quản trong lọ thủy tinh màu. Dung dịch B: thêm 120ml amoni hydroxid đặc vào 330ml nước cất. Khi dùng trộn hai dung dịch theo tỷ lệ tương đương về thể tích.
Dung dịch natri acetat TT.218
Dung dịch natri acetat (NaCH3COO) trong nước nồng độ 13,6% m/v (~1N)
Dung dịch natri azid TT.219
Dung dịch natri azid trong nước nồng độ 5% m/v.
Dung dịch natri bisulfit TT.220
Pha dung dịch natri bisulfit trong nước nồng độ ~0,5N. Kiểm tra pH nếu cần. Điều chỉnh pH về 3,0-4,0 bằng acid sulfuric loãng hoặc natri hydroxid.
Dung dịch natri bitartrat TT.221
Dung dịch natri bitartrat (NaHC4H4O6.H2O) trong nước nồng độ ~1N. Pha dung dịch ngay trước khi sử dụng.
Dung dịch natri borat TT.222
Dung dịch natri borat (Na2B4O7.10H2O) trong nước nồng độ 2% m/v.
Dung dịch natri carbonat TT.223
Dung dịch natri carbonat khan (Na2CO3) trong nước nồng độ 10,6% m/v.
Dung dịch natri clorid TT.224
Dung dịch natri clorid trong nước nồng độ 10% m/v.
Dung dịch natri cobal tnitrit TT.225
Hòa tan 10g natri cobaltnitrit [Na3Co(NO2)] trong nước, định mức đến 50ml. Lọc nếu cần
Dung dịch natri ethoxid TT.226
Hòa tan 10g natri trong 120ml etanol tuyệt đối theo cách sau: loại bỏ màng dầu bảo vệ trên bề mặt của natri bằng giấy lọc, cân natri trong dung dịch benzol sau đó lau khô benzol bằng giấy lọc. Cắt miếng natri ra nhiều mảnh nhỏ, cẩn thận cho từng miếng vào bình cầu chứa 120ml etanol, bình cầu được nối với sinh hàn hồi lưu.
Dung dịch natri fluorescen TT.227
Dung dịch natri fluorescen trong etanol 50% nồng độ 0,1% m/v.
Dung dịch natri florid TT.228
Làm khô 0,5g natri florid trong 4 giờ tại 2000C. Cân chính xác 0,222g natri florid khan, hòa tan trong nước cất, định mức đến 100ml. Hút 1ml dung dịch này cho vào bình định mức dung tích 1000ml, định mức đến vạch bằng nước cất, lắc đều. 1ml dung dịch này tương ứng với 0,01mg Flo (F).
Dung dịch natri hydro sulfit TT.229
Dung dịch natri hydro sulfit trong nước nồng độ 33,3%. Pha dung dịch ngay trước khi sử dụng.
Dung dịch natri hydroxid TT.230
Hòa tan 4,3g natri hydroxid trong nước cất, định mức đến 100ml (nồng độ ~1N)
Dung dịch natri hydroxid trong metanol TT.231.
Hòa tan 5g natri hydroxid trong 5ml nước cất, thêm metanol đến đủ 100ml, gạn lấy phần dung dịch trong.
Dung dịch natri nitrit TT.232
Dung dịch Natri nitrit trong nước nồng độ 10% m/v. Pha dung dịch ngay trước khi sử dụng.
Dung dịch natri nitroferricyanid (dung dịch natri nitroprussid) TT.233
Dung dịch Natri nitroferricyanid [Na2Fe(NO)(CN)5.2H2O] trong nước nồng độ 5% m/v. Pha dung dịch ngay trước khi sử dụng.
Dung dịch natri phosphat dibasic (dung dịch natri phosphat) TT.234
Dung dịch natri phosphat dibasic (pha từ tinh thể trong -Na2HPO4.7H2O) trong nước nồng độ 12% m/v.
Dung dịch natri phosphat monobasic TT.235
Dung dịch Natri phosphat monobasic (NaH2PO4.2H2O) trong nước nồng độ 62,4% m/v (~4M).
Dung dịch natri starch glycolat (5%) TT.236
Tẩm ướt 5g natri starch glycolat bằng vài giọt etanol. Thêm 100ml nước, đun sôi trong khoảng 2 - 3 phút, để nguội.
Dung dịch natri starch glycolat (1%) TT.237
Lấy 10ml dung dịch natri starch glycolat (5%) TT.236 pha loãng đến 50ml bằng nước cất. Pha dung dịch ngay trước khi sử dụng.
Dung dịch natri sulfid TT.238
Dung dịch natri sulfid (Na2S.9H2O) trong nước nồng độ 10% m/v. Pha dung dịch ngay trước khi sử dụng.
Dung dịch natri sulfit (PbT) TT.239
Hòa tan Natri sulfit (PbT) trong nước cất, định mức đến 100ml, lọc.
Dung dịch natri thiosulfat TT.240
Dung dịch natri thiosulfat trong nước nồng độ 0,1N.
Dung dịch thiếc clorid TT.241
Hòa tan 3,2g thiếc clorid (SnCl2.2H2O) trong 40ml dung dịch acid hydroclorid 0,3N. Chuyển dung dịch này vào bình định mức 100ml, định mức đến vạch bằng acid hydroclorid 0,3N. Pha dung dịch ngay trước khi sử dụng. Trước khi sử dụng dung dịch thiếc clorid cần được chuẩn lại bằng dung dịch acid sulfuric periodic TT.247, điều chỉnh để 10ml dung dịch thiếc clorid chuẩn với 10,2ml dung dịch acid periodic. Trong quá trình chuẩn độ cần cho thêm 5ml acid hydroclorid đặc và 1ml chỉ thị hồ tinh bột vào 10ml dung dịch thiếc clorid (dung dịch chuyển thành màu xanh tại điểm tương đương).
Dung dịch tinh bột (Hồ tinh bột) TT.242
Tán nhỏ 1g tinh bột trong 10ml nước lạnh. Rót chậm, khuấy đều vào trong 200ml nước sôi. Đun sôi hỗn hợp đến khi trở thành dung dịch hồ loãng (đun lâu quá sẽ làm giảm độ nhạy). Để dung dịch ổn định, sử dụng phần dung dịch trong phía trên. Pha dung dịch trước khi sử dụng.
Dung dịch acid sulfanilic TT.243
Dung dịch acid sulfanilic (p-NH2.C6H4SO3H.H2O) trong acid acetic nồng độ 0,8%. Bảo quản trong lọ kín
Dung dịch acid sulfanilic/α-naphtylamin TT.244
Hòa tan 0,5g acid sulfanilic (p-NH2.C6H4SO3H.H2O) trong 150ml acid acetic. Hòa tan 0,1g α-naphtylamin trong 0,26g acid hydrocloric và 150ml acid acetic. Trộn hai dung dịch này. Khi để lâu dung dịch này xuất hiện màu hồng thêm bột kẽm để loại màu.
Dung dịch acid sulfuric TT.245
Pha loãng thể tích acid sulfuric đã biết trước nồng độ với nước cất để thu được dung dịch acid sulfuric có nồng độ H2SO4 94,5-95,5%
Dung dịch acid sulfuric loãng TT.246
Dung dịch chứa khoảng 10% H2SO4 m/v. Cách pha: thêm cẩn thận 57ml H2SO4 nồng độ 95-98% hoặc Dung dịch acid sulfuric TT.245 vào khoảng 100ml nước cất, làm mát dung dịch về nhiệt độ phòng, sau đó pha loãng với nước cất đến 1000ml (~2N).
Dung dịch acid sulfic/acid periodic TT.247
Hòa tan 3,42g acid periodic (H5IO6) trong 100ml dung dịch acid sulfuric 0,25M. Chuyển dung dịch này vào bình định mức dung tích 500ml, định mức đến vạch bằng acid sulfuric 0,25M. (~0,03M acid sulfuric/acid periodic).
Dung dịch acid tanic TT.248
Hòa tan 1g acid tanic (tannin) trong 1ml etanol, thêm nước cất đến đủ 10ml. Pha dung dịch ngay trước khi sử dụng.
Dung dịch acid tanic/acid acetic băng TT.249
Hòa tan 10g acid tanic (tannin) trong 80ml acid acetic băng, lắc đều. Thêm 32ml acid phosphorric. Pha dung dịch ngay trước khi sử dụng.
Dung dịch tartrat kiềm TT.250
Hòa tan 34,6g kali natri tartrat (muối Rochelle) và 10g natri hydroxid trong nước cất, định mức đến đủ 100ml. Để yên 2 ngày, lọc qua bông thủy tinh.
Dung dịch thymol xanh TT.251
Hòa tan 0,1g thymol xanh trong 100ml etanol, lọc nếu cần. Để làm chi thị xác định pH hòa tan 0,1g thymol xanh trong 4,3ml natri hydroxid 0,05N, pha loãng và định mức bằng nước cất không có carbon dioxid đến 200ml.
Dung dịch thioure TT.252
Dung dịch thioure trong nước nồng độ 10% m/v.
Dung dịch thymolphtalein TT.253
Hòa tan 0,1g thymolphtalein trong 100ml etanol, lọc nếu cần.
Dung dịch thiếc (II) sulfat TT.254
Hòa tan 10g thiếc (II) sulfat trong 100ml acid sulfuric 1%. Khuấy dung dịch liên tục trong vài giờ, gạn lấy dịch trong.
Dung dịch trinitrophenol (dung dịch acid picric) TT.255
Hòa tan khối lượng trinitrophenol tương đương với 1g trinitrophenol khan trong 100ml nước cất nóng. Làm nguội dung dịch và lọc nếu cần.
Dung dịch uranyl acetat TT.256
Hòa tan 1g uranyl acetat trong 20ml nước cất. Lắc kỹ hỗn hợp và lọc.
Dung dịch uranyl kẽm acetat TT.257
Hòa tan 10g uranyl acetat [(CH3COO)2UO2.2H2O] bằng cách đun với 50ml nước cất và 5ml dung dịch acid acetic trong nước (30% m/v; ~5N). Hòa tan 10g kẽm acetat [Zn(CH3COO)2] với 30ml nước cất và 3ml dung dịch acid acetic trong nước (30% m/v; ~5N). Trộn đều hai dung dịch, để nguội về nhiệt độ phòng, lọc loại bỏ phần không tan.
Dung dịch acid vanadic/acid molybdic TT.258
Hòa tan 1,12g amoni metavanadat trong khoảng 300ml nước ấm và 250ml acid nitric, để nguội dung dịch. Hòa tan 27g amoni molybdat trong khoảng 400ml nước cất ấm. Trộn đều hai dung dịch này. Thêm nước tới đủ 1000ml. Bảo quản dung dịch trong chai thủy tinh màu, dung dịch dùng trong 3-4 ngày kể từ khi pha.
Dung dịch xylenol da cam TT.259
Dung dịch xylenol da cam trong etanol nồng độ 0,1% m/v.
Dung dịch kẽm amalgam (hỗn hống) TT.260
Cho khoảng 10g bột kẽm vào khoảng 20ml thuỷ ngân để tạo ra dung dịch amalgam lạnh, đun nóng đến 1500C, khuấy liên tục đến khi kẽm tan hết. Kẽm amalgan có thể tái sử dụng cho đến khi hàm lượng kẽm còn khoảng 0,2% m/m. Hàm lượng kẽm được xác định như sau: Cho đầy thủy ngân vào pinomet tại 250C±10C, cân picnomet (mHg), tiến hành tương tự đối với amalgan (mAmg), tính hàm lượng kẽm:
Hàm lượng kẽm % m/m =
Trong đó:
A =
Dung dịch kẽm sulfat TT.261
Dung dịch kẽm sulfat (ZnSO4.7H2O) trong nước nồng độ 10% m/v.
Dung dịch kẽm sulfat chuẩn TT.262
Hòa tan 0,44g kẽm sulfat trong nước cất, định mức đến đủ 1000ml. Lấy 50ml dung dịch này, định mức bằng nước cất đến 1000ml. 1ml dung dịch chứa 5μg Zn.
Dung dịch zirconi/alizarin TT.263
Dung dịch A: hoà tan 0,8g zirconi nitrat [Zn(NO)3.5H2O] trong nước cất, thêm vài giọt acid nitric 4N, định mức đến 100ml bằng nước cất. Dung dịch B: hòa tan 0,1g alizarin sulfonat monohydrat trong 20ml nước cất, định mức đến 100ml bằng etanol. Trộn 1ml dung dịch A, 1ml dung dịch B và 18ml nước cất. Dung dịch này phải trong. Quá trình pha loãng dung dịch nên tiến hành ngay trước khi sử dụng.
Dung dịch Zwikker TT.264
Trộn 1ml pyridin với 4ml dung dịch đồng sulfat (10% trong nước) và 5ml nước cất.
Dung dịch metyl tím (dung dịch tím tinh thể, metyl rosanilin clorid) TT.265
Dung dịch metyl tím trong acid acetic băng nồng độ 1%.
Dung dịch acid hydrocloric TT.266
Dung dịch acid hydrocloric trong nước nồng độ 35% m/v.
- 1Quyết định 3005/QĐ-BYT năm 2014 công bố kết quả hệ thống hóa văn bản quy phạm pháp luật về y tế tính đến ngày 31 tháng 12 năm 2013 do Bộ trưởng Bộ Y tế ban hành
- 2Thông tư 08/2015/TT-BYT sửa đổi Thông tư 27/2012/TT-BYT hướng dẫn việc quản lý phụ gia thực phẩm do Bộ Y tế ban hành
- 3Công văn 1758/ATTP-KN năm 2015 về phụ gia thực phẩm Lecithin do Cục An toàn thực phẩm ban hành
- 1Nghị định 86-CP quy định phân công trách nhiệm quản lý nhà nước về chất lượng hàng hoá
- 2Quyết định 14/1999/QĐ-TTg về việc thành lập Cục Quản lý chất lượng vệ sinh an toàn thực phẩm thuộc Bộ Y tế do Thủ tướng Chính phủ ban hành
- 3Nghị định 49/2003/NĐ-CP quy định chức năng nhiệm vụ, quyền hạn và cơ cấu tổ chức của Bộ Y tế
- 4Thông tư 08/2015/TT-BYT sửa đổi Thông tư 27/2012/TT-BYT hướng dẫn việc quản lý phụ gia thực phẩm do Bộ Y tế ban hành
- 5Công văn 1758/ATTP-KN năm 2015 về phụ gia thực phẩm Lecithin do Cục An toàn thực phẩm ban hành
Quyết định 4021/2003/QĐ-BYT ban hành "Quy định đánh giá vệ sinh an toàn 20 chất phụ gia thực phẩm" do Bộ trưởng Bộ Y tế ban hành
- Số hiệu: 4021/2003/QĐ-BYT
- Loại văn bản: Quyết định
- Ngày ban hành: 30/07/2003
- Nơi ban hành: Bộ Y tế
- Người ký: Trần Chí Liêm
- Ngày công báo: Đang cập nhật
- Số công báo: Từ số 469 đến số 470
- Ngày hiệu lực: 08/09/2008
- Ngày hết hiệu lực: 01/08/2011
- Tình trạng hiệu lực: Kiểm tra